Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Palabras clave: adrenal, high blood pressure, pheochromocytoma
El presente artículo busca describir las manifestaciones clínico–humorales ante una paciente con feocromocitoma. Es por ello que se presenta el caso de una paciente de 52 ańos que acude a la institución por presentar aumento de sus indicadores tensionales acompańado de frialdad de la piel, sudoraciones, nerviosismo, dolor torácico opresivo que aparece varias veces en el día, pérdida de peso en torno a los 13 kilos y decaimiento. Lo más significativo en los estudios realizados fue el ultrasonido abdominal que definió una imagen nodular en la suprarrenal derecha de baja ecogenicidad y contornos regulares que miden 5 mm, confirmada con tomografía axial computarizada contrastada de suprarrenales. Se determinó ácido vanilmandélico hallándose valores aumentados. Asimismo, la biopsia de glándula suprarrenal derecha confirmó la presencia de feocromocitoma de 4,5 x 3,5 x 3 cm.
En el polo superior de ambos riñones se localizan las glándulas suprarrenales o adrenales. Ellas son aplanadas con forma de media luna y un tamaño que puede variar según edad y condiciones fisiopatológicas, pero por lo general ambas pesan ocho gramos. Desde el punto de vista histológico existe una corteza y una médula [1],[2],[3].
El feocromocitoma es una neoplasia neuroendocrina o tumor de la médula adrenal, de células cromafines derivadas de la cresta neural que biosintetiza, almacena y secreta catecolaminas, fundamentalmente adrenalina, noradrenalina y, en menores cantidades, dopamina. Los tumores desarrollados sobre restos del tejido cromafín son menos frecuentes y se pueden encontrar en el cuello, mediastino posterior, órgano de Zuckerkandl, pelvis, vejiga urinaria, saco pericárdico, encéfalo y discos dermoides. Diversos autores los denominan paragangliomas funcionantes, reservando el nombre de feocromocitoma para los que se originan en la médula [1],[2],[3],[4],[5] .
El feocromocitoma diagnosticado precozmente evita la muerte súbita y permite curar la hipertensión arterial que produce, no dejando secuelas en órganos diana como el corazón riñón y retina [1],[2],[3],[4],[5] . Por estas razones reportamos el caso clínico, para describir las manifestaciones clínico-humorales de una paciente con feocromocitoma.
Paciente femenina de 52 años, raza blanca, con antecedente de hipertensión arterial de 13 años de evolución, que ha llevado varios tratamientos antihipertensivos sin lograr control de las cifras tensionales. Al ingreso tenía tratamiento con enalapril, nifedipina y clortalidona. Dos meses antes de su ingreso presentó dolor retroesternal opresivo acompañado de piel fría, sudoración, palpitaciones, decaimiento, pérdida de alrededor de 13 kilos de peso, cefalea de localización occipital de moderada intensidad, hipertensión paroxística sistólica y diastólica que oscilaba en valores de 240 mmHg de sistólica y 130 mmHg de diastólica.
Los estudios realizados presentaron los siguientes valores: hemoglobina 13,1 g/l; leucocitos 9,7 x 109/l (linfocitos 16,0%, monocitos 0,5%, neutrófilos 69,7%, eosinófilos 8,4%, basófilos 0,9%); conteo de plaquetas 307 x 109/l; eritrosedimentación 4 mm/h; glicemia 11,2 mmol/l; creatinina 114 mmol/l; colesterol 6,56 mmol/l; triglicéridos 10,54 mmol/l; ácido úrico 349 mmol/l; fosfatasa alcalina 201 UI; TGP 44 UI; triyodotironina 3,2 nmol/l y tiroxina130 nmol/l.
La radiografía de tórax posteroanterior registró un área cardíaca normal, sin alteraciones pleuropulmonares.
El fondo de ojo presentó un ligero estrechamiento arteriolar que puede ser observado en la hipertensión arterial, pero no se corresponde con retinopatía hipertensiva, que ya sería un daño de la retina y por tanto de órgano diana.
En los estudios bioquímicos la determinación de metanefrina en orina arrojó ácido vanilmandélico en valores superiores a 10 mg% y con dos determinaciones en valores superiores a 15 mg%.
Dentro de los estudios de localización tumoral, el ultrasonido abdominal presentó hígado con aumento difuso de su ecogenicidad que rebasa en 2 cm el reborde costal, con vesícula acodada de paredes finas con varias imágenes litiásicas en su interior. En la proyección de la glándula suprarrenal derecha y en contacto con el lóbulo derecho, la imagen nodular es de baja ecogenicidad y contornos regulares que miden 5 mm. La tomografía axial computarizada contrastada de suprarrenales mostró imagen hiperdensa que capta contraste hasta 112 UH en su periferia, con zona central más hiperdensa 63 UH a nivel de suprarrenal derecha que en fase venosa se capta de forma más homogénea, pero con zonas de menos densidad por necrosis. Ello impresiona por relacionarse con feocromocitoma de suprarrenal derecha. El estudio anatomopatológico posterior a la adrenalectomía derecha describió células que varían de forma, citoplasma granular basófilo, núcleos pleomórficos de cromatina laxa, nucléolos prominentes en nidos separados por tabiques fibrovasculares con signos de benignidad (véase Figura 1).
Figura 1. Feocromocitoma de glándula suprarrenal derecha.
Las manifestaciones del exceso de catecolaminas (adrenalina y noradrenalina), la sudoración, taquicardia, dolor retroesternal opresivo, epigastralgia, cefalea de moderada intensidad y la hipertensión arterial paroxística; sumado a los datos del examen físico como piel fría, sudorosa, ruidos cardiacos taquicárdicos, cifras tensiónales elevadas, dos determinaciones elevadas de metanefrina en orina (ácido vanilmandélico); más los estudios imagenológicos de localización tumoral como el ultrasonido abdominal que muestra imagen nodular de baja ecogenicidad de suprarrenal derecha; la tomografía axial computarizada contratada de suprarrenales que confirma la existencia de imagen hiperdensa de suprarrenal derecha; y el estudio anatomopatológico posterior a la adrenalectomía derecha con signos de benignidad; confirman el diagnóstico positivo de feocromocitoma benigno esporádico de suprarrenal derecha.
Al presentar hipertensión paroxística, el tratamiento específico del caso clínico se realizó con fentolamina (amp 10 mg) en dosis de 1-5 mg en bolo endovenoso cada 5 minutos hasta controlar las cifras tensionales. Para el manejo preoperatorio se tuvo en cuenta complicaciones como crisis hipertensivas durante la inducción anestésica y fluctuaciones extremas de la presión sanguínea durante la manipulación del tumor. Por estas razones a la paciente se le preparó varios días antes del tratamiento quirúrgico. Al principio se utilizó alfa-bloqueadores que provocan vasodilatación, disminuyendo las cifras tensionales. Luego se trató con beta-bloqueadores para controlar la taquicardia y combatir las arritmias que provocan los alfa-bloqueadores. No se retiró del tratamiento el anti-cálcico, porque la asociación con el alfa-bloqueador controla adecuadamente las cifras tensionales. Esto no ocurría así cuando la paciente lo empleaba con otros antihipertensivos antes del diagnóstico de feocromocitoma.
El tratamiento con la fenoxibenzamina (alfa-bloqueador) se comenzó con 10 mg cada 12 horas, aumentando de 10-20 mg cada cuatro días hasta llegar a 80 mg en total. El propanolol (beta-bloqueador) se suministró a razón de 10 mg cada 8 horas y el nifedipino (anti-cálcico) a razón de 30 mg diarios.
En las 12 horas previas a la cirugía se administró infusión intravenosa de 2.000 ml de solución salina fisiológica al 0,9%, para evitar hipotensión severa tras la resección.
La evolución posterior a la adrenalectomía derecha fue favorable, dado que se controló la hipertensión arterial, no presentó hipotensión arterial y las manifestaciones de la descarga adrenalítica desaparecieron, motivo por el cual la paciente no continúa con tratamientos antihipertensivos. A los 15 días de la cirugía las metanefrinas mostraron valores normales. El caso se siguió al primer, tercer y sexto mes en consultas de medicina interna y endocrinología. La paciente se encuentra asintomática, siendo el pronóstico favorable.
El feocromocitoma es un tumor benigno en 90% de los casos, sólo el 10% es maligno, 10% aparece en la infancia, 10% de presentación familiar (regla de los 10) y 90% es esporádico. Representa entre 0,1 y 1% de las causas de hipertensión arterial secundaria. No obstante, se desconoce su incidencia real por tener formas asintomáticas como es el caso de los incidentalomas suprarrenales (5 a 6,5%) y diagnosticarse en 0,1% en necropsias. Por lo general, 90% es unilateral y 10% bilateral [1],[8],[3]. Este tumor no tiene predilección por sexo ni edad, aunque se ha visto con mayor frecuencia entre la cuarta y quinta décadas de vida. Su tamaño varía entre 5 y 6 mm de diámetro y pueden pesar desde 50 o 200 g hasta varios kilos.
Según la literatura consultada, entre 90 y 98% se localiza en el sector abdominal. Se caracteriza por una producción excesiva de catecolaminas como la adrenalina, noradrenalina y, en menor cantidad, dopamina. Estas sustancias, al ser liberadas al torrente sanguíneo, ocasionan una serie de manifestaciones clínicas entre las que predominan las alteraciones cardiocirculatorias, hipertensión arterial mantenida en 25% de los casos, o como crisis hipertensivas, y disminución del volumen sanguíneo. Si el diagnóstico no se realiza en forma precoz, puede generar complicaciones severas como la miocardipatía dilatada, infarto del miocardio y hemorragia cerebral, a pesar de que el feocromocitoma sea benigno.
Durante las crisis el paciente puede presentar cefalea, frialdad, palidez, sofocación facial, temblor, ansiedad, palpitaciones, náuseas, diarreas, dolor torácico o abdominal y parestesias en las extremidades. Al finalizarlas puede generar postración con marcada debilidad muscular. Estas manifestaciones pueden aparecer espontáneamente o por ejercicios físicos, cambios de postura, exposición al frío, defecación, micción, coito o palpación abdominal [1],[2][3].
Este tumor puede dar lugar a procesos metabólicos, hipercalcemia, hipertiroidismo, síndrome de Cushing, diabetes mellitus por disminución del nivel de insulina y por el exceso de catecolaminas, llamadas hormonas contrainsulares. También puede ocasionar trastornos de la esfera psiquiátrica como neurosis de ansiedad e incluso puede ser causa de muerte súbita [2],[3],[4].
El feocromocitoma puede encontrarse en 90% de los casos asociado a enfermedades neuroectodérmicas como neurofibromatosis tipo 1 o Von Reckinghausen (su diagnóstico se establece al tener dos o más de los siete criterios: seis o más manchas color café con leche, dos o más neurofibromas cutáneos o neurofibromas plexiformes, neurofibromas inguinales, pecas en la región axilar, un glioma en el nervio óptico, familiares en primer grado con neurofibromatosis tipo 1 o asociado a enfermedad de Von Hippel Lindau –quistes del sistema nervioso central, hemagioblastoma de la retina y tumores benignos en diversos órganos-), Sturger Weber (ataxia cerebelosa, esclerosis tuberosa), carcinoma medular del tiroides, hiperparatiroidismo y astrocitoma cerebral o formar parte de los síndromes de adenomatosis endocrino-múltiple II o síndrome de Sipple (feocromocitoma, carcinoma medular de tiroides, hiperparatiroidismo y con frecuencia es bilateral multicéntrico y maligno) o adenomatosis endocrino-múltiple III o IIb (feocromocitoma, carcinoma medular de tiroides, neuromas mucosos y hábito marfanoide) [5],[6],[7],[8],[9].
En la literatura se reportan casos clínicos de pacientes con incidentalomas suprarrenales con características morfológicas, parámetros clínicos y de laboratorio que han sugerido feocromocitoma. Ejemplo de ello es el caso de una paciente femenina de 35 años, portadora de un tumor adrenal izquierdo por estudio ecográfico, a quien se le realizó adrenalectomía que confirmó el diagnóstico. Otro caso clínico es el de una paciente femenina de 47 años de edad, asintomática y sin antecedentes patológicos conocidos. Durante la evaluación preoperatoria para trasplante renal de donante vivo, la angiografía por tomografía evidenció una tumoración en glándula suprarrenal izquierda. Se concluyó evaluación pre trasplante sin ninguna contraindicación por lo cual se decidió realizar adrenalectomía izquierda, existiendo feocromocitoma de 4 x 3 cm [10].
La etiología incluye tumores benignos y malignos de todas las zonas de la corteza y de médula suprarrenal, metástasis hacia las glándulas suprarrenales, así como enfermedades infiltrativas. Ambos casos clínicos presentaron evolución satisfactoria, al igual que el caso reportado [10].
Otro caso mostrado es el de una familia con enfermedad de Von Hippel Lindau, donde uno de los miembros de sexo masculino, de 52 años, además presentó feocromocitoma bilateral con hemangioblastoma de la retina, quien falleció súbitamente por enfermedad cerebral vascular. Comparado con otros casos presentados, su evolución no resultó favorable [9].
Otro caso da cuenta de un paciente joven portador de un feocromocitoma, que sometido a la larga acción de las catecolaminas llegó a desarrollar una miocardiopatía dilatada con criterio de trasplante cardiaco. En él, la cirugía adrenal fue curativa [11].
Por último, se presenta un paciente de 32 años cuyo padre hipertenso fallece a los 38 años sin causa precisa. El paciente ingresa por el debut de una hipertensión paroxística, se demuestra por ultrasonido y tomografía axial computarizada la existencia de grandes tumores suprarrenales bilaterales compatibles con feocromocitoma. El paciente fallece cuatro días después del ingreso debido a un edema agudo del pulmón, en el curso de una emergencia hipertensiva. El diagnóstico de feocromocitoma se comprueba por anatomía patológica, tanto macroscópica como microscópica [3].
Con el reporte de estos casos clínicos se confirmó que el feocromocitoma no tiene predilección por el sexo, aparece entre la cuarta y quinta décadas de la vida y puede constituir un hallazgo en estudios imageneológicos comportándose como incidentaloma. Con frecuencia se presenta de manera esporádica y a veces asociado a enfermedades genéticas. Evoluciona favorablemente y en algunos casos existen complicaciones que llevan al paciente a la muerte, a pesar del diagnóstico temprano.
La hipertensión idiopática o esencial es frecuente en 95% de los casos, donde existen con mayor frecuencia manifestaciones clínicas de cefalea de localización occipital, zumbido de los oídos, dolor precordial y sudoraciones. A veces es asintomática. Sin embargo, no se demuestra la presencia de tumoración suprarrenal y humoralmente los valores de metanefrinas en orina como el ácido vanilmandélico y otras catecolaminas en plasma, son normales [10],[11],[12].
Debido al estado hipercinético de los pacientes con feocromocitoma, debe diferenciarse del hipertiroidismo. Clínicamente presentan taquicardia, sudoraciones y pérdida de peso, pero las determinaciones hormonales de tiroxina y tetrayodotironina, resultaron normales y la hormona tiro-estimulante del tiroides no mostró supresión de sus valores como expresión de un hipertiroidismo subclínico o manifiesto [13],[14],[15].
La diabetes mellitus tipo 2 es de comienzo insidioso a partir de la cuarta década de la vida, con manifestaciones clínicas como la sed intensa, poliuria, polidipsia y la determinación en plasma de valores glicémicos en ayunas o sin ella por encima de los 11,1 mmol/l. La prueba de tolerancia a la glucosa presenta valores en ayunas por debajo de 7,8 mmol/l, a las 2 horas por encima de 11,1 mmol/l o más, y dos determinaciones de glicemia en ayunas por sobre 7 mmol/l. En el caso presentado la diabetes es secundaria a una enfermedad endocrina productora de catecolaminas y se confirmó con una determinación de la glicemia en ayunas en 11,2 mmol/l.
El hiperaldosteronismo primario presenta como manifestación patognomónica hipertensión arterial moderada, no paroxística ni intermitente, con presencia de un tumor de la corteza adrenal, pero los valores de catecolaminas en plasma y orina son normales.
Puede sospecharse psiconeurosis pues las manifestaciones de sudoración, taquicardia, ansiedad están presentes en estos casos, pero no existe presencia de un tumor. En los antecedentes del caso reportado no se hace referencia a trastorno de esfera psíquica.
En situaciones especiales como el estrés grave, cirugía antes del diagnóstico del feocromocitoma, cetoacidosis, infarto cerebral, uso de medicamentos como el paracetamol, antidepresivos tricíclicos, puede existir hipersecreción de catecolaminas sin presencia de tumor de las suprarrenales [12],[13],[14],[15]. Estas situaciones no se reportan en la paciente.
En la literatura consultada el pronóstico es favorable, con una supervivencia en los casos benignos de 95% a los 5 años, con recidivas entre 5 y 10% [12]. Se reporta que el seguimiento debe efectuarse a los 15 días con determinación de catecolaminas al primer mes y luego a los tres, seis y 12 meses siguientes y de manera anuales durante cinco años [12],[13],[14],[15].
En la literatura revisada los pacientes con manifestaciones clínicas más frecuentes por hipersecreción de catecolaminas (hipertensión arterial intermitente o mantenida, diaforesis, cefalea, sudoraciones, taquicardia, pérdida de peso), se les indica programa de estudio para diagnóstico de feocromocitoma. Dentro de estos tenemos los estudios bioquímicos de alta sensibilidad como la determinación plasmática de metanefrinas (99%), seguidas de las metanefrinas urinarias fraccionadas (97%) y determinación menos sensible es el ácido vanilmandélico (64%). Cuando no puedan efectuarse estos análisis, se indican estudios de localización tumoral como tomografía axial computarizada, resonancia magnética nuclear (sensibilidad de 93 a 100%) y ultrasonido de suprarrenales que presenta menor sensibilidad (64%)[12],[13],[14],[15].
Otros estudios reportados en la literatura son las pruebas funcionales de imágenes, como la gammagrafía con 123 I-metayodobencilguanidina, de elección. En su defecto puede realizarse con 131 I-metayodobencilguanidina. Ambas presentan sensibilidad y especificidad del 100% para la localización de feocromocitoma y permiten identificar malignidad o multicentricidad del tumor. Se basan en el hecho de que las células del feocromocitoma expresan sistema de transporte de membrana y vesiculares de catecolaminas, lo que permite imagen con 131 I-metayodobencilguanidina y 123 I-metayodobencilguanidina [12],[13].
Otros estudios diagnósticos de importancia son los genéticos, debido a que existen formas familiares con herencia autosómica dominante con mutación en el gen VHL o en el gen que codifica las subunidades B y D de la succionado deshidrogenasa, como MEN II a y MEN II b: mutación en protooncogen RET. En ellos el 50% presentan feocromocitoma y Von Hippel-Lindau, implicado el gen VHL. Entre 10 y 20 % desarrollan feocromocitoma. En otras neurofibromatosis como el Von Reklinghausen existe feocromocitoma entre 0,1 y 5,7%.
Ante un paciente con feocromocitoma esporádico se debe realizar estudio genético, sobre todo si el paciente diagnosticado es menor de 21 años, si el tumor es bilateral o extraadrenal o existen paragagliomas múltiples, los cuales desarrollan feocromocitoma en el 20% de los casos [12],[13],[14],[15].
Dentro de esta supresión con clonidina, las pruebas dinámicas son de gran importancia para detectar los falsos positivos en las determinaciones plasmáticas de metanefrinas y catecolaminas. La clonidina es un agonista alfa-2 de acción central, que en personas normales disminuye las concentraciones de catecolaminas. La persistencia de estos metabolitos confirma feocromocitoma [13],[14],[15].
Confirmado el feocromocitoma, el tratamiento plantea dos momentos. En la fase previa a la adrenalectomía debe realizarse el manejo preoperatorio y transoperatorio, con el objetivo de disminuir la hipertensión arterial y aumentar el volumen sanguíneo. Al inicio es necesario usar alfa-bloqueadores que provocan vasodilatación preoperatoria con reexpansión del volumen contraído del plasma, disminuyendo la tensión arterial y el efecto soporífero. Así el paciente llega relajado y apacible a la sala de anestesia. El empleo de beta-bloqueadores debe ejecutarse ser exclusivamente cuando se logre el bloqueo alfa-adrenérgico, con el objeto de inhibir los efectos de las catecolaminas sobre la actividad del miocardio. El alfa-bloqueador de mayor uso es la fenoxibenzamina (20 a 100 mg, 3 veces al día por vía oral). Otro fármaco de este grupo con buenos resultados es la doxazosina (2 a 8 mg al día), que pueden disminuir la desventaja de los alfa-bloqueadores que producen arritmia.
Logrado el bloqueo alfa-adrenérgico, se continúa con beta-bloqueadores como el propanolol (10 mg, 3 veces al día, vía oral) en los casos de hipertensión mantenida. El labetalol, un fármaco con acción antagonista alfa, puede utilizarse en una dosis entre 200 y 600 mg cada 12 horas, pero su acción alfa-bloqueadora es insuficiente por lo que no es de elección.
El tratamiento quirúrgico de los feocromocitoma puede ser por vía laparoscópica si se trata de tumores menores de 8 cm, de presentar mayor tamaño este método presenta desventajas respecto de la cirugía convencional. La cirugía presenta complicaciones perioperatorias dada la manipulación del tumor, lo que provoca crisis hipertensivas y arritmias. Las crisis hipertensivas que tienen lugar durante la cirugía se tratan con nitroprusiato sódico (potente vasodilatador arterial y venoso), por su acción rápida a razón de 0,5 y 1 g/kg/min, dosis que se ajusta según las cifras de tensión arterial. Con nitroprusiato se produce un aumento reflejo de la frecuencia cardiaca, por lo que hasta 60% de los pacientes requieren añadir beta-bloqueadores durante la cirugía, frente a 20% cuando se emplean anti-cálcico [12],[13],[14],[15].
Declaración de conflictos de intereses
Los autores han completado el formulario de declaración de conflictos de intereses del ICMJE traducido al castellano por Medwave, y declaran no haber recibido financiamiento para la realización del reporte; no tener relaciones financieras con organizaciones que podrían tener intereses en el artículo publicado, en los últimos tres años; y no tener otras relaciones o actividades que podrían influir sobre el artículo publicado. El formulario puede ser solicitado contactando a la autora responsable.
Comité de ética
El presente manuscrito fue evaluado y aprobado por el comité de ética institucional correspondiente.
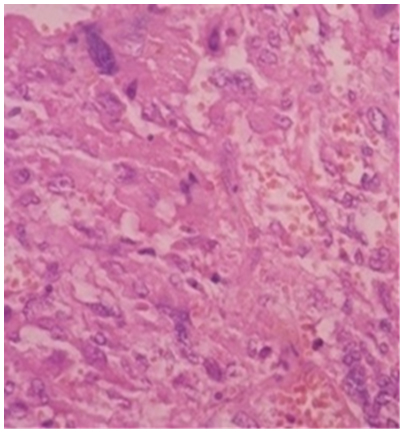 Figura 1. Feocromocitoma de glándula suprarrenal derecha.
Figura 1. Feocromocitoma de glándula suprarrenal derecha.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
The article aims to describe the clinical and laboratory features of a female patient suffering from pheochromocytoma. The case is a 52-year-old female patient who presents to our healthcare center with high blood pressure, cold limbs, sweating, jitteriness, and episodes of oppressive chest pain that appear several times per day. She also reports fatigue and a 13-kilogram weight loss. The sonogram revealed a nodular image in the right adrenal gland that had low echogenicity and regular margins measuring 5 mm. The image was confirmed with a contrast-enhanced adrenal CAT scan. Urine vanillylmandelic acid levels were high and an adrenal biopsy confirmed a pheochromocytoma measuring 4.5 x 3.5 x 3 cm.
Autores:
Otmara Aranguren Barreto[1], Idania Teresa Mora López[1], Maritza Cardosa Samón[1], Olga León González[1], María Victoria López Soto[1], Ramón Portales Rodríguez Pérez[1]
Citación: Aranguren O, Mora IT, Cardosa M, León O, López MV, Portales R. Clinical and laboratory features of pheochromocytoma in a 52-year-old female patient. Medwave 2014;14(3):e5942 doi: 10.5867/medwave.2014.03.5942
Fecha de envío: 21/1/2014
Fecha de aceptación: 1/4/2014
Fecha de publicación: 22/4/2014
Origen: no solicitado
Tipo de revisión: con revisión por tres pares revisores externos, a doble ciego
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Junquera LC, Carneiro J. Glándulas endócrinas. En: Texto y atlas de histología
básica. 4ed. Barcelona: Masson, 1996:373-98. Gurwitz J. Trastornos suprarrenales. Sección 12: enfermedades endocrinas y
metabólicas. En: Manual Merck de diagnóstico y tratamiento. Espańa: Elsevier,
2007:1300-1317. Arocha Molina Y, Acosta Piedra Y, Piedra Herrera B, Suárez Díaz T, Madruga Vázquez K. Feocromocitoma bilateral: presentación de un caso. Rev Med Electron. 2011;33(2):239-243. | Link |Cabrera Gámez M, Silva Turcios T, Fuentes M, González Calero T, Yanes Quesada M, Díaz Socorro C. Feocromocitoma: presentación de un caso clínico. Rev Cubana Endocrinol. 2008;19(2) | Link |Parrilla Delgado ME, López Soto MV, Valls Pérez O, Borrajero Martínez I, Rubio N,
Cubero Rego D, et al. Atlas de ecocitopatología diagnóstica en las lesiones abdominales. La Habana: Editorial de Ciencias Médicas, 2006:229-32. Botella JI, Valero MA, Timón IM, Álvarez F, García G, Luque M, et al. Patologías
suprarrenales. En: Manual de diagnóstico y terapéutica en endocrinología y
nutrición. Servicio de Endocrinología, Hospital Ramón y Cajal, 2007:103-129. Marcos Carlos A. Feocromocitoma: presentación de un caso. Rev Argent Ultrason. 2012;11(1):28-30. Cadena M, Vergara A, Olarte A, Ospina D. Paraganglioma del órgano de Zuckerkandl. Rev Colomb Cir. 2010;25(4):309-322. | Link |Salman P, Vucetich N, López JM. Enfermedades de von Hippel Lindau en una familia chilena: diagnóstico clínico y genético. Rev Chil Endocrinol Diabetes. 2010;3(1):19-23. | Link |Franceschi KJ, Guerrero G, Fontana N, Aceveo O, Alemán L. Incidentaloma en evaluación pre trasplante de donante vivo. Rev Venez Oncol. 2011;23(1):42-45. | Link |González González JL, Menéndez Núńez J, Escarpanter JC, Abela Lazo A. Miocardiopatía dilatada y feocromocitoma. Rev Cubana Cir. 2013;52(1). | Link |Oleaga A, Gońi F. Feocromocitoma: actualización diagnóstica y terapéutica. Endocrinol Nutr. 2008;55(5):202-16. | PubMed |Arteaga Hernández JL, Plaza González T, Suero Almonte S, Calzadilla García L, Almora Carbonel CL. Feocromocitoma. Presentación de un caso. Rev Cienc Med. 2011;15(3):197-204. | Link |Dieguez A. Academia Nacional de Medicina. Consenso Nacional Inter-Sociedades para el diagnóstico y tratamiento de las neoplasias renales parenquimatosas del adulto. Rev Arg Radiol. 2011;74(3). | Link |
Incidentaloma suprarrenal: żpor qué y cuándo operar?






 Estudios originales
Estudios originales