Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en reunión clínica del Departamento de Medicina, Hospital Clínico Universidad de Chile. El director del Departamento de Medicina es el Dr. Alejandro Cotera y la coordinadora de las reuniones clínicas es la Dra. Miriam Alvo.
La masa suprarrenal clínicamente inaparente se define como una masa suprarrenal que se descubre incidentalmente en un examen de imagen realizado durante el estudio de una condición clínica no relacionada con la sospecha de enfermedad suprarrenal, excluyendo aquellos casos en que el estudio ha tenido como objetivo la etapificación y seguimiento de un cáncer. De ahí la denominación de incidentaloma suprarrenal, cuya frecuencia ha ido en aumento a medida que ha aumentado el número y sensibilidad de los exámenes radiológicos, especialmente en relación a la masificación del uso de la Tomografía Axial Computada (TAC).
La frecuencia de incidentaloma suprarrenal es cercana a 6% en las autopsias, con rango entre 1 y 32%; en exámenes radiológicos como el ultrasonido disminuye a menos de 1% y en el caso de la TAC de abdomen se encuentra esta masa en 1,3% de los casos, pero es probable que esta cifra aumente debido a la solicitud creciente de TAC ante cualquier tipo de malestar abdominal. El hallazgo es más frecuente a mayor edad del paciente: en menores de 30 ańos su frecuencia es menor de 1%, mientras que en los mayores de 70 ańos puede alcanzar hasta 7%.
Las masas suprarrenales clínicamente inaparentes pueden corresponder a: adenomas suprarrenales, feocromocitomas, mielolipomas, ganglioneuromas, quistes, hematomas, cánceres o metástasis.
En la Tabla I se muestra la frecuencia de algunos incidentalomas evaluados en la Clínica Mayo (1). Como se observa, la gran mayoría corresponde a tumores benignos o adenomas, no funcionantes.
Tabla I. Características histológicas y bioquímicas de incidentalotas suprarrenales.
En el caso del adenoma productor de cortisol, la gran mayoría determina un Síndrome de Cushing subclínico es decir, los pacientes tienen aumento de la secreción de cortisol, pero no presentan el fenotipo clásico del síndrome de Cushing.
Sólo 10 a 15% de los incidentalomas son bilaterales. La gran mayoría de estas lesiones son nódulos de menos de tres centímetros de diámetro. En la revisión de Barzon, se demostró que la funcionalidad o exceso de secreción hormonal se relaciona directamente con el tamańo de los tumores. Los nódulos mayores de 4 cm tienen mayor probabilidad de ser hiperfuncionantes alcanzando casi el 50% en los mayores de 6 cm (2).
Entre 5% y 25% de los incidentalomas aumentan de tamańo (al menos un centímetro) durante el seguimiento. El riesgo de crecer aumenta dependiendo del tiempo de evolución: al ańo el riesgo acumulativo es 6% y a los cinco ańos, 30%. La tasa de malignización oscila entre 1/4000 hasta 1/1000 y es mayor en pacientes de sexo masculino. La posibilidad global de que estos tumores se conviertan en tumores funcionantes es de 20%; como ya se mencionó, el riesgo depende directamente del tamańo del tumor y es mucho más probable en tumores mayores de tres centímetros en el momento del diagnóstico. En estos casos el riesgo acumulativo es de 17% al ańo y puede llegar hasta 47% a los cinco ańos de seguimiento (3). En una serie de 75 pacientes con una masa suprarrenal asintomática que fueron seguidos hasta por 15 ańos, el riesgo de desarrollar hiperfunción fue 4% al ańo y alrededor de 10% a los cinco ańos, manteniéndose estable hasta los 10 ańos de seguimiento (4).
La mayoría de los incidentalomas suprarrenales que inicialmente son funcionantes o que se transforman en funcionantes durante su evolución, son secretores de cortisol (5). La probabilidad de que un incidentaloma no funcionante en el momento del diagnóstico produzca un síndrome de Cushing clínico es de alrededor de 7% a los cinco ańos de seguimiento; si en el momento del diagnóstico el paciente ya presenta un hipercortisolismo subclínico, la probabilidad de que desarrolle un Cushing clínico al ańo sube a 12,5%. Esto es importante de tener en cuenta al momento de tomar una decisión terapéutica.
La TAC y la Resonancia Magnética (RM) son igualmente efectivas para el diagnóstico y diferenciación entre lesiones benignas y malignas, pero es muy importante considerar la calidad del examen y de su informe. Un aspecto importante en el caso de la TAC es que el examen se realice con medio de contraste, para evitar que la superposición de estructuras provoque falsos positivos. No hay evidencia suficiente que respalde la utilidad de los exámenes cintigráficos, de modo que no son de primera línea en el estudio de estos pacientes. La punción aspirativa tiene alto porcentaje de falsos negativos, ya que la citología de la glándula suprarrenal es bastante confusa en su interpretación, a diferencia de la citología de la glándula tiroides; por lo tanto tampoco se utiliza como primera línea de estudio para evaluar el tipo de masa suprarrenal. Puede ser útil en pacientes con historia de cáncer y sospecha de metástasis en la suprarrenal.
En 2002, el National Institutes of Health (NIH) organizó un consenso de expertos para que aportara recomendaciones sobre el manejo del incidentaloma suprarrenal, con base en los datos disponibles hasta ese momento. En este consenso se recomendó descartar funcionalidad mediante: prueba de supresión nocturna con 1 mg de dexametasona; medición de metanefrinas fraccionadas en muestra de orina o libres en plasma; y en el caso de pacientes hipertensos, medición del nivel de potasio y de la relación entre aldosterona y actividad de renina plasmática (6).
El punto de corte en el test de supresión con dexametasona, o test de Nugent, corresponde a cortisol plasmático menor de 1,8 ug/dl. Antiguamente se utilizaba como punto de corte una concentración menor de 5 ug/dl, pero con éste se obtenía 20% de falsos negativos. En el ańo 2003, en un consenso para el diagnóstico del síndrome de Cushing se estableció el valor actual, que tiene como desventaja que aumenta los falsos positivos. En el Laboratorio de Endocrinología del Hospital Clínico de la Universidad de Chile (HCUCH) se realizó una evaluación del test de Nugent en un grupo de pacientes con depresión, que pueden cursar con alteraciones en la supresión con dexametasona sin padecer el síndrome de Cushing. Los resultados se compararon con los de un grupo control sin depresión. En el grupo con depresión, 1 paciente de 38 (2,6%) tuvo cortisol mayor de 1,8 pero bajo 3 ug/dl; en el grupo control, 1 paciente de 40 (2,5%) presentó niveles en esos rangos. Estos resultados sugieren que si se obtiene un valor sobre 1,8 ug/dl en el test de supresión con dexametasona no se debe catalogar de inmediato el paciente como portador de síndrome de Cushing. Por el contrario, se debe realizar todo un protocolo de estudio que incluye repetir el test de Nugent y realizar otras pruebas. Para establecer el diagnóstico de Síndrome de Cushing subclínico deben existir dos o más pruebas alteradas de las que clásicamente se realizan en el estudio de un síndrome de Cushing suprarrenal: ACTH suprimida, pérdida de ritmo circadiano de cortisol, DHEASO4 suprimida y alteraciones del cortisol libre en orina de 24 horas. Aquí también se puede considerar el uso del cintigrama con 131I-Norcolesterol o 131I-6 beta-Iodometilnorcolesterol.
En la interpretación del valor de cortisol plasmático se debe considerar que lo que se mide es el cortisol unido a su proteína transportadora, por lo tanto todos los factores que aumenten esta proteína, como el uso de estrógenos orales en anticonceptivos o la terapia hormonal de reemplazo, pueden ocasionar falso aumento de los valores; asimismo, las condiciones de estrés, como hospitalización o enfermedades agudas y algunos fármacos que aumentan el metabolismo de la dexametasona, como rifampicina y los anticonvulsivantes: fenitoína, fenobarbital y carbamazepina también pueden causar falsos positivos en el test de Nugent.
Para el diagnóstico de feocromocitoma se debe efectuar determinación de epinefrina, norepinefrina, metanefrina y normetanefrina en orina de 24 horas. La medición de ácido vainillil mandélico en orina se ha ido dejando de lado, por su baja sensibilidad.
Frente a la sospecha de feocromocitona lo primero es verificar que el paciente no esté recibiendo fármacos que puedan alterar el estudio, como antihipertensivos alfa y beta-bloqueadores o clonidina; inhibidores de la monoaminooxidasa, incluyendo algunos antidepresivos, cafeína y los antiparkinsonianos, entre otros. Además, los pacientes deben suprimir alimentos que contengan vainillín y ácido fenólico, excepto si la medición se realiza con cromatografía líquida de alta eficiencia (HPLC), en la cual estos factores dietéticos no interfieren. Por último, estas mediciones también pueden aumentar falsamente en pacientes con insuficiencia renal.
Se ha planteado que las metanefrinas serían más útiles que las catecolaminas para el diagnóstico de feocromocitoma. Estas últimas dependen directamente de la secreción que tenga el tumor en un momento determinado, mientras que la metabolización de catecolaminas a metanefrinas es un proceso más lento, por lo tanto su medición no refleja sólo un momento en la secreción del tumor, sino la acumulación de este producto en el tiempo por lo que tiene mayor sensibilidad. Bravo y su grupo evaluaron la utilidad de estos exámenes y observaron que tanto en feocromocitomas hereditarios: síndrome de neoplasia endocrina múltiple tipo II, síndrome de von Hippel Lindau, como en los casos esporádicos, la medición de metanefrinas urinarias fraccionadas (metanefrina y normetanefrina) tiene muy buena sensibilidad, 96 a 97% pero una baja especificidad en casos esporádicos (45 %). En cuanto a las catecolaminas fraccionadas en orina (epinefrina y norepinefrina), son menos sensibles en casos hereditarios (79%) y menos específicos en casos esporádicos (75%), por lo que algunos grupos postulan que para mejorar la sensibilidad y la especificidad se debe realizar las dos mediciones en forma simultánea (7). En nuestro país es difícil lograr este objetivo por el elevado costo, de modo que en nuestro grupo se ha optado por la medición de las metanefrinas, teniendo precaución con los usuarios crónicos de benzodiazepinas, ya que si estos fármacos se han suspendido recientemente se puede producir aumento de los niveles de metanefrinas asociado al efecto de la privación aguda.
El grupo de Bravo ha demostrado que la medición de metanefrinas libres en plasma, tiene la mejor sensibilidad y especificidad y con este examen se evitaría la recolección de orina en 24 horas y sería de gran utilidad en nińos o pacientes con insuficiencia renal.
Se debe sospechar hiperaldosteronismo en pacientes hipertensos con o sin hipokalemia. En pacientes normotensos la frecuencia de hiperaldosteronismo es <1%. (H. Vierhapper. Exp Clin Endocrinol Diabetes 2007; 115: 518 – 521).
En los hipertensos se debe solicitar la medición de aldosterona y actividad de renina plasmática (ARP); la relación aldosterona/ARP igual o superior a 30 se considera diagnóstica de hiperaldosteronismo. La muestra para este examen se toma antes de las 9 de la mańana, luego de que el paciente haya estado al menos dos horas levantado y por lo menos quince minutos sentado; estos detalles son muy importantes para que el resultado sea confiable. Aunque algunos grupos plantean que no es indispensable, para aumentar la sensibilidad de esta relación, previo a la toma del examen se debería tratar la hipokalemia, indicar una dieta libre en sodio, suspender la espironolactona y cualquier tipo de fármaco que interfiera con el eje renina-angiotensina-aldosterona. Idealmente estos pacientes hipertensos sólo deberían estar recibiendo alfa-bloqueadores en el momento del estudio, porque la gran mayoría de los antihipertensivos puede alterar esta relación y producir falsos positivos o falsos negativos, como se puede observar en la Tabla II.
En el último tiempo se ha demostrado que algunos adenomas e hiperplasias macronodulares bilaterales ACTH independiente productores de cortisol, son regulados por la expresión aberrante en la célula de la zona fascicular de la corteza suprarrenal de receptores ectópicos (8). Se trata de receptores diferentes al de la hormona adrenocorticotrópica (ACTH), pero que también están acoplados a proteína G y activan la vía esteroidogénica cuando se unen a sus ligandos naturales (catecolaminas, GIP (polipéptido inhibitorio gástrico o polipéptido insulinotrópico dependiente de glucosa), vasopresina, hormona luteinizante (LH), gonadotropina coriónica, serotonina, angiotensina) aumentando la secreción de cortisol.
La presencia de estos receptores ectópicos se debe a mutaciones somáticas producidas en el período postcigótico o embrionario; su estimulación determina la proliferación monoclonal o policlonal, determinando el crecimiento de un adenoma unilateral o de una hiperplasia macronodular bilateral.
Como ejemplo de éstos tenemos el síndrome de Cushing dependiente de alimentos, que se caracteriza por niveles de cortisol normales en ayunas y elevados en el estado post prandial, y el síndrome de Cushing dependiente de LH, que se manifiesta en mujeres durante el embarazo, para luego ceder y reaparecer en la menopausia.
Lo interesante en esta patología, es que algunos de estos pacientes podrían recibir tratamiento médico; por ejemplo, si se demuestra estimulación por presencia de receptores beta-adrenérgicos, se podrían tratar con beta-bloqueadores. Por esto, cuando existe sospecha de alguno de estos cuadros se debe aplicar un protocolo de estudio que incluya todos los estímulos para precisar los posibles receptores involucrados y así establecer la etiología de los nódulos y de la hipersecreción hormonal (9).
Actualmente se considera que la indicación quirúrgica es clara cuando existe evidencia clínica de funcionalidad, evidencia bioquímica de feocromocitoma o cuando el tumor, aunque no sea funcionante, tiene más de 6 centímetros. Estos criterios también se establecieron en el consenso ya mencionado (6). Se estableció como punto de corte 6 cm, ya que el riesgo de malignidad en estos casos es de 25%, mientras que en tumores menores de 4 cm el riesgo es menor de 2%.
En el caso de los pacientes sin criterios quirúrgicos se recomienda hacer seguimiento clínico y con imágenes cada seis meses durante los primeros tres ańos, ya que durante este periodo los tumores tienen mayor probabilidad de crecer y hacerse funcionantes; luego el paciente se debe controlar en forma anual. No se ha establecido el tiempo total de seguimiento: algunos expertos seńalan que luego de cinco ańos sin crecimiento ni evidencia de funcionalidad se podría dar de alta al paciente, pero esto es discutible porque luego de diez ańos un tumor todavía puede crecer y/o volverse maligno. Por otra parte, es lógico suponer que los pacientes con tumores de tamańo límite, entre tres y cinco centímetros y los pacientes jóvenes deberían tener un seguimiento mucho más prolongado.
En un seguimiento de cinco ańos efectuado en el Hospital Clínico de la Universidad de Chile en pacientes con hallazgo de nódulo suprarrenal en una TAC de abdomen realizada como parte del estudio de algún problema extrarrenal o como control postquirúrgico, excluyendo a los casos con diagnóstico de tumor primario de otro origen, se realizó una evaluación hormonal en todos los pacientes para descartar funcionalidad. El tiempo de seguimiento fluctuó entre seis meses y cinco ańos. Se incluyó a 22 pacientes, 14 mujeres y 8 hombres con 55 ańos de edad promedio. Las características de los casos se resumen en la Tabla III:
Como consecuencia de los resultados de esta serie se decidió establecer, en nuestro centro, que había en todo nódulo mayor de 3 cm, a pesar de que el consenso internacional fija el criterio en 6 cm o más.
 Tabla I. Características histológicas y bioquímicas de incidentalotas suprarrenales.
Tabla I. Características histológicas y bioquímicas de incidentalotas suprarrenales.
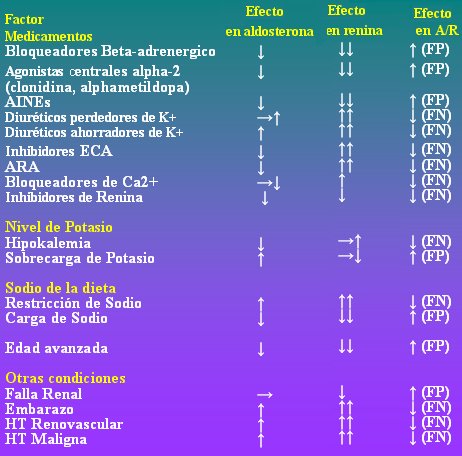 Tabla II. Efectos de diferentes situaciones clínicas en la relación Aldosterona/ARP. FP: falso positivo. FN: falso negativo. Adaptado de Funder JW et al. J Clin Endocrinol Metab. 2008 Sep
Tabla II. Efectos de diferentes situaciones clínicas en la relación Aldosterona/ARP. FP: falso positivo. FN: falso negativo. Adaptado de Funder JW et al. J Clin Endocrinol Metab. 2008 Sep
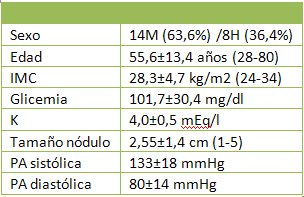 Tabla III. Características de 22 pacientes con incidentaloma suprarrenal. Hospital Clínico de la Universidad de Chile 2000-2005.
Tabla III. Características de 22 pacientes con incidentaloma suprarrenal. Hospital Clínico de la Universidad de Chile 2000-2005.
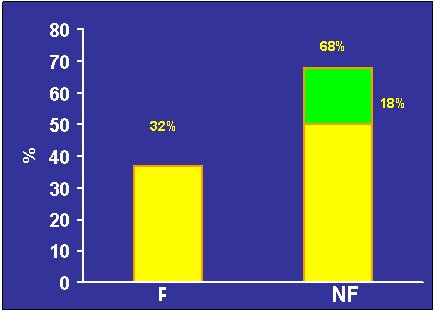 Figura 1. Resultados observados en 22 pacientes con incidentaloma suprarrenal. Hospital Clínico de la Universidad de Chile 2000-2005.
Figura 1. Resultados observados en 22 pacientes con incidentaloma suprarrenal. Hospital Clínico de la Universidad de Chile 2000-2005.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en reunión clínica del Departamento de Medicina, Hospital Clínico Universidad de Chile. El director del Departamento de Medicina es el Dr. Alejandro Cotera y la coordinadora de las reuniones clínicas es la Dra. Miriam Alvo.
Autora:
Verónica Araya[1]
Citación: Araya V. Clinically inapparent adrenal mass. Medwave 2009 Ago;9(8):e4076 doi: 10.5867/medwave.2009.08.4076
Fecha de publicación: 1/8/2009
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Young WF Jr. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007 Feb 8;356(6):601-10. | CrossRef | PubMed |Barzon L, Sonino N, Fallo F, Palu G, Boscaro M. Prevalence and natural history of adrenal incidentalomas. Eur J Endocrinol. 2003 Oct;149(4):273-85. | CrossRef | PubMed |Libč R, Dall'Asta C, Barbetta L, Baccarelli A, Beck-Peccoz P, Ambrosi B. Long-term follow-up study of patients with adrenal incidentalomas. Eur J Endocrinol. 2002 Oct;147(4):489-94. | CrossRef | PubMed |Barzon L, Scaroni C, Sonino N, Fallo F, Paoletta A, Boscaro M. Risk factors and long-term follow-up of adrenal incidentalomas. J Clin Endocrinol Metab. 1999 Feb;84(2):520-6. | CrossRef | PubMed |Barzon L, Fallo F, Sonino N, Boscaro M. Development of overt Cushing's syndrome in patients with adrenal incidentaloma. Eur J Endocrinol. 2002 Jan;146(1):61-6. | CrossRef | PubMed |Grumbach MM, Biller BM, Braunstein GD, Campbell KK, Carney JA, Godley PA, et al. Management of the clinically inapparent adrenal mass ("incidentaloma"). Ann Intern Med. 2003 Mar 4;138(5):424-9. | PubMed |Bravo EL, Tagle R. Pheochromocytoma: state-of-the-art and future prospects. Endocr Rev. 2003 Aug;24(4):539-53. | CrossRef | PubMed |






 Estudios originales
Estudios originales