Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Palabras clave: diabetic nephropathy, microvascular complications, chronic renal disease physiopathology
La enfermedad renal crónica es una complicación frecuente en la diabetes mellitus. Su importancia radica en la alta prevalencia y la proyección a futuro que ésta tiene. Se asocia a altos gastos en salud y además a deterioro cardiovascular global. La fisiopatología del desarrollo de esta enfermedad está siendo estudiada y se sabe que en ella participan una serie de vías moleculares complejas que determinan una enfermedad microvascular. En esta revisión se intenta abordar las vías conocidas en el desarrollo de nefropatía diabética, con el fin de comprender mejor posibles blancos terapéuticos que se podrían desarrollar.
La diabetes es una enfermedad crónica, que se caracteriza por la incapacidad del páncreas para producir insulina en cantidades suficientes o por el ineficiente uso de esta. Tomando en cuenta la importancia que tiene esta hormona para el control del azúcar en la sangre, una falla en su producción y regulación produce progresivamente un daño grave en órganos y sistemas [1],[2],[3].
Se estima que existen al menos 284 millones de personas diabéticas en el mundo, los que se proyectan a 436 millones para el año 2030 [2],[3]. En tanto en Chile, la Encuesta Nacional de Salud mostró una prevalencia total de 9,4% [4].
Las complicaciones de la diabetes pueden ser macro y micro vasculares. Éstas aumentan la probabilidad de padecer enfermedades cardiovasculares, acrecentando el riesgo de muerte en dos veces comparado con población sin la patología [1],[2],[3],[4],[5].
La fisiopatología de la nefropatía diabética no está completamente clara, aunque se sabe que las principales noxas serían el aumento descontrolado de la glicemia, especialmente cuando se acompaña de hipertensión arterial. Existen algunos factores de riesgo para desarrollar nefropatía, entre los cuales se destacan la historia familiar de enfermedad renal o de hipertensión, un mal control glicémico, diabetes tipo 1 antes de los 20 años y el hábito tabáquico [3].
Antiguamente se consideraba a la nefropatía como una enfermedad no inmunológica, pero estudios han demostrado que los procesos inflamatorios e inmunitarios están involucrados en la patogenia de esta enfermedad [6],[7]. Además, en el desarrollo de la misma están incluidas muchas vías que conllevan a la injuria renal, como son productos avanzados de la glicosilación y vía de los polioles, estrés oxidativo, factores de crecimiento profibróticos y citoquinas que promueven inflamación, entre otros [6],[7].
El objetivo de esta revisión es comprender los mecanismos que son responsables del daño microvascular renal. Se describirán los procesos patológicos que ocurren a nivel glomerular en la diabetes.
Se realizó una revisión bibliográfica en PubMed, Scholar Google, documentos oficiales de la Organización Mundial de la Salud y documentos del Ministerio de Salud de Chile. Como palabras clave se utilizaron pathophyisiology, diabetic nephropathy, microvascular complications, advanced glycation end products, oxidative stress, aldose reductase, renineangiotensine aldosterone system, protein kinase c, inflammation citokines, growth factors, gene, epigenetic, miRNAs.
Se seleccionaron inicialmente 53 estudios, de los cuales se descartaron 15 por no cumplir los criterios de inclusión. Dentro de los 38 seleccionados había estudios de cohorte, de intervención, de prevalencia, de casos y controles y estudios genéticos que contuvieran las palabras claves en relación al desarrollo de nefropatía diabética.
Definición de nefropatía diabética
La nefropatía diabética es una complicación causada por la diabetes a nivel de la microvasculatura renal. Los individuos con diabetes presentan una mayor tasa de filtración glomerular o hiperfiltración, mediada por la mayor relajación de las arteriolas aferentes en comparación a las eferentes. A su vez, esto conduce a un aumento del flujo sanguíneo a través del capilar glomerular, elevando la presión. Cuando estas condiciones se mantienen en el tiempo, producen tanto una hipertrofia glomerular como un aumento de la superficie del capilar glomerular. Ello causa alteraciones hemodinámicas que contribuyen al desarrollo y/o progresión de esta enfermedad [8],[9].
Se han identificado tres fases capilares en el desarrollo de la nefropatía diabética:
1.- Fase capilar normal: las células mesangiales están normalmente montadas sobre los capilares del glomérulo. Cuando esta célula se contrae, tracciona la membrana basal y reduce el diámetro de los capilares.
Para entender esto se deben conocer los elementos que determinan la tasa de filtración glomerular afectada por las leyes de Starling, en donde la presión hidrostática mueve el líquido hacia la cápsula de Bowman. Además, el coeficiente de reflexión de las proteínas es prácticamente uno y el ultrafiltrado está libre de éstas, por lo que la presión oncótica en el capilar es prácticamente cero. Por lo tanto, la presión hidrostática del capilar es la única fuerza que favorece la filtración. La presión hidrostática en el espacio de Bowman y la presión oncótica en el capilar se oponen a la filtración. Además, el descenso de la resistencia de arteriola aferente aumenta la filtración, mientras que un aumento de resistencia la reduce. Por el contrario, una disminución en la resistencia en la arteriola eferente disminuye la filtración y el aumento en la resistencia aumenta la filtración [10].
Este efecto reduce la presión hidrostática y, por consiguiente, la filtración glomerular también disminuye. La contracción de las células mesangiales está mediada por angiotensina II que actúa sobre receptores angiotensina I en la membrana de estas células [11]. Además, las células mesangiales tienen la función de sintetizar matriz y degradar matriz envejecida [12].
2.- Fase de hiperfiltración/microalbuminuria: la hiperglicemia disminuye la contractilidad de las células mesangiales, debido a que esta condición de glucosa sanguínea elevada favorece la glicosilación de las fibras de F-actina en la célula mesangial [13]. Por lo tanto, ocurre un aumento del diámetro capilar. Además, se ha demostrado que los pacientes diabéticos tienen una respuesta aumentada de vasoconstricción postglomerular producto de la angiotensina II [14]. Estos cambios se traducen en un aumento de presión en el glomérulo y en hiperfiltración. También, en esta etapa de desarrollo comienza la acumulación de lámina densa y matriz mesangial [11].
3.- Fase de macroalbuminuria e insuficiencia renal: se llega a esta fase si la hiperglicemia persiste por años. Consecuentemente la célula mesangial se expande más y se sigue acumulando matriz y lámina densa. Estos cambios producen que el capilar glomerular sea aplastado por la célula mesangial y desencadenan insuficiencia renal [11].
En resumen, las lesiones más tempranas consisten en engrosamiento de la membrana glomerular basal, expansión mesangial y acumulación hialina en las arteriolas. La nefropatía ya establecida se caracteriza por la expansión mesangial nodular, acumulación hialina en arteriolas aferente y eferente, y una membrana basal del glomérulo marcadamente engrosada. Además la perdida de los podocitos es crucial en la esclerosis del glomérulo [15].
La nefropatía diabética se clasifica por su daño intersticial y vascular, según los hallazgos en la biopsia y la microscopia de luz:
Figura 1. Factores que participan en el desarrollo de nefropatía diabética.
Los mecanismos que se abordaran en esta revisión serán:
1) Productos avanzados de glicosilación
En la diabetes, el estado persistente de hiperglicemia genera glicosilación proteica no enzimática reversible. Estos productos glicosilados pueden reorganizarse en otros cada vez más estables, como por ejemplo la hemoglobina glicosilada, hasta que se vuelven irreversibles mediante una sucesión de reordenamientos en su estructura. Estos últimos productos irreversibles se denominan productos finales avanzados de glicosilación, los que se acumulan por una hiperglicemia mantenida en el tiempo. Además, han sido ligados a consecuencias renales [16],[17]. Se han encontrado receptores para los productos avanzados de glicosilación en diferentes zonas del riñón. Estos se encuentran en células endoteliales, mesangiales, tubulares y musculares lisas [17]. Cuando se ligan estos productos finales con sus respectivos receptores se produce liberación de citoquinas proinflamatorias, factores de transcripción, factores de crecimiento y, además, se observa activación de vías de señalización. Todo esto permite la generación de especies reactivas de oxígeno, daño endotelial, activación de procesos inflamatorios y, por ende, daño microvascular en los riñones.
2) Estrés oxidativo
Este estado de desbalance causado por la hiperglicemia sostenida tiene varias fuentes, tanto no enzimáticas como enzimáticas. Ejemplos de la primera son los productos de la glicosilación, vía de señalización de polioles, auto-oxidación de la glucosa y alteraciones en el metabolismo mitocondrial. Por otro lado, en las fuentes enzimáticas son importantes las enzimas nicotinamida adenina dinucleótido fosfato oxidasa y enzimas antioxidantes, ambas alteradas en el desarrollo de estrés oxidativo. Se ha visto que en la nefropatía diabética existe un aumento del estrés oxidativo en todas las estructuras del riñón [16],[17]. Los radicales libres principalmente tienen el objetivo de causar una peroxidación en los lípidos de membrana, con lo que causan una pérdida de integridad y disfunción de las membranas. Además, el estrés oxidativo altera el funcionamiento del glomérulo debido al daño que produce en las células mesangiales y en las células del endotelio glomerular [18],[19].
3) Vía de polioles
La vía de los polioles es aquella implicada en la formación de sorbitol. Esta tiene inicio en la acción de la enzima aldosa reductasa, que convierte irreversiblemente glucosa en sorbitol, sobre el que luego actúa sorbitol deshidrogenasa para producir fructosa. Normalmente, la hexoquinasa transforma glucosa en glucosa-6-fosfato, pero en condiciones de hiperglicemia esta enzima se satura, por lo que se produce mayor cantidad de sorbitol y hay también un mayor consumo de nicotinamida adenina dinucleótido fosfato por parte de la aldosa reductasa, sugiriendo como posibles mecanismos de daño la falta de poder reductor para apalear el estrés oxidativo y el edema producido por la mayor concentración del sorbitol [20].
Existen estudios que demuestran mayor expresión de la aldosa reductasa en pacientes con nefropatía diabética, de modo que se ha encontrado una actividad significativamente mayor en diabéticos con nefropatía, respecto a diabéticos sin nefropatía [20].
Un estudio en ratas mostró que un inhibidor de la aldosa reductasa prevenía los efectos de la nefropatía diabética de modo que reduce la sobreexpresión renal del factor de crecimiento endotelial vascular [21]. Otro estudio en ratas reveló que la enzima RSOR/MIOX (Renal-specific oxido-reductase/myoinositol oxigenase), que cataboliza la producción de mioinositol y presenta ciertas similitudes con la aldosa reductasa, también estaría implicada en la producción de daño a nivel renal. Esto porque se encontró una relación directa entre niveles aumentados de ROSOR/MIOX y fibronectina tubuloinsterticial, factor de crecimiento transformante β y proteína kinasa C, entre otros. Además, fue posible observar que al inhibir la aldosa reductasa se revertían parcialmente estos cambios [22].
4) Papel de la proteína kinasa C
En diabéticos, se activan vías de señalización como proteína kinasa C, incrementando la producción de especies reactivas del oxígeno y, por consiguiente, desencadenando el estrés oxidativo que daña la macro y micro circulación en el organismo. Esta vía depende de la nicotinamida adenina dinucleótido fosfato oxidasa, sobre expresada en las células mesangiales del riñón en pacientes diabéticos, generando un daño localizado en este órgano [23]. Por otro lado, la generación de isoformas de la proteína kinasa C y los altos niveles de diacilglicerol sanguíneo conducen a la disfunción microvascular y a la progresión de la enfermedad renal crónica. También, se le atribuyen otros efectos en su sobreexpresión, como la apoptosis y proliferación celular, contribuyendo de manera independiente al daño renal [24].
Entre los daños que genera esta vía de señalización se encuentran alteraciones en la permeabilidad renal (albuminuria), alteraciones en la tasa de filtrado glomerular, fibrosis del tejido intersticial, generación de especies reactivas del oxígeno y engrosamiento de la membrana basal. Todo esto altera la función renal pudiendo generar insuficiencia renal crónica a largo plazo [25].
5) Sistema renina angiotensina aldosterona
El sistema renina angiotensina en la nefropatía diabética se encuentra activado por la proteinuria. La angiotensina II promueve la producción de factores quimiotácticos y moléculas de adhesión, la formación de citoquinas fibrogénicas y desregulación en la producción y degradación de matriz extracelular, cambios que conllevan a una esclerosis del glomérulo. Se han encontrado los niveles de angiotensina II elevados, producto de la proteinuria y especies reactivas del oxígeno [26].
En el caso de los podocitos, estos presentan acumulaciones de proteínas oxidadas y mitocondrias dañadas, ya que la angiotensina II induce la formación de especies reactivas del oxígeno, favoreciendo la autofagia de estas células, produciendo un aumento de la filtración glomerular [27]. En la actualidad, los tratamientos están enfocados a bloquear la angiotensina II en cualquiera de sus etapas [28].
6) Factores de crecimiento
Entre los factores de crecimiento importantes en la nefropatía diabética están factor de crecimiento transformante β, factor de crecimiento de tejido conectivo, factor de crecimiento endotelial vascular y el factor de crecimiento tipo insulina 1 [7],[14].
El factor de crecimiento transformante β está expresado débilmente en un glomérulo renal normal. En la diabetes, la hiperglicemia, proteína kinasa C, productos avanzados de glicosilación, especies reactivas del oxígeno y angiotensina II estimulan la expresión de éste, principalmente en células renales, mesangiales y del túbulo contorneado proximal. Este factor de crecimiento es importante en el desarrollo de la nefropatía ya que promueve tejido fibrótico en las células túbulo-instersticiales mediante la acumulación de colágeno, fibronectina y laminina. Ello inhibe la acción de colagenasas y disminuye la acción del sistema inmunológico [7],[14].
El factor de crecimiento de tejido conectivo es otro participante de la patogenia de la nefropatía como respuesta a la expresión del factor de crecimiento transformante β. También aumenta su expresión en la hiperglicemia celular, estrechamiento mecánico de la arteriola eferente renal y especies reactivas del oxígeno que producen factor de crecimiento de tejido conectivo directamente. Ambos factores, inducen la producción de matriz extracelular, proliferación celular, supervivencia y adhesión. La mayor expresión de factor de crecimiento de tejido conectivo induce a la activación de factor de crecimiento transformante β, produciendo retroalimentación positiva para la producción y acumulación de tejido fibrótico en células renales [7].
El factor de crecimiento endotelial vascular es importante en la producción de nuevos vasos sanguíneos, crecimiento de vasos ya existentes y, por consiguiente, en el desarrollo de la nefropatía diabética, aumentando su expresión por el factor de crecimiento transformante β y angiotensina II. Este factor se encuentra principalmente en podocitos, túbulos colectores y túbulos contorneados distales. Ha sido encontrado en hipertrofia glomerular y renal, así como en la hiperfiltración como respuesta a la diabetes [7].
El factor de crecimiento de tipo insulina-1 está mediado por un receptor de membrana tipo tirosina kinasa, participando en la hipertrofia y aumento de la producción de matriz extracelular. El descontrol de la glicemia por la diabetes disminuye la producción de éste en el hígado, lo que produce una disminución del factor en la sangre. En respuesta a ello, se genera una estimulación local de síntesis de este factor (por inducción de la hipersecreción de la hormona del crecimiento) en tejidos no hepáticos, entre ellos, el riñón [9].
7) Citoquinas inflamatorias
Inicialmente la nefropatía se consideraba como una patología no inmune. Sin embargo, hoy en día existen evidencias para responsabilizar a la inflamación y a las respuestas inmunes en la aparición y desarrollo de esta enfermedad [29]. Estudios recientes demuestran que las citoquinas inflamatorias son determinantes en la aparición de las complicaciones microvasculares como la neuropatía, retinopatía y la nefropatía diabética [30]. Además, el estado de hiperglicemia se asocia a la activación de mecanismos que conllevan a la producción de citoquinas, aumentando el daño inflamatorio [31].
Debido a la acción sinérgica en ocasiones de las citoquinas, la respuesta inflamatoria se refuerza y se hacen más notorios los efectos. Por otro lado, las citoquinas también incitan la expresión de otras citoquinas y de receptores de las mismas. En la actualidad se reconocen a la interleuquina-1, interleuquina-6, interleuquina-8 y el factor de necrosis tumoral como actores del desarrollo de la nefropatía diabética y como potenciadores de las complicaciones en esta enfermedad [32].
Al factor de necrosis tumoral se le asocia con la retención de sodio e hipertrofia renal y como marcador temprano de la nefropatía diabética [33]. Por otra parte, se ha encontrado que la interleuquina-18 estimula la formación de otras citoquinas, como la interleuquina-1 y el factor de necrosis tumoral [34], contribuyendo al deterioro de las células renales, su posterior daño y el desarrollo de la nefropatía diabética. A su vez, actúa como factor quimiotáctico para neutrófilos y monocitos, incrementando la respuesta inflamatoria exagerada a nivel del intersticio renal y promoviendo el daño del mismo [22].
En el caso de la interleuquina-1, se ha encontrado sobre expresada en pacientes con nefropatía diabética [35], junto con la expresión de los factores quimiotácticos y las moléculas de adhesión [36]. Además, la interleuquina-1 aporta en el desarrollo de anormalidades en la hemodinámica intraglomerular y la síntesis de prostaglandinas por parte de las células mesangiales. Por otro lado, la expresión de genes de la ciclooxigenasa-2, fosfolipasa A, óxido nítrico inducible, está mediada por la interleuquina-1. Finalmente, todo lo anterior está asociado a la acumulación de matriz extracelular en las células mesangiales, aumentando la presión en el capilar glomerular [31].
8) Epigenética
La susceptibilidad de desarrollar cambios epigenéticos varía según el momento de la vida. Es así como el tiempo comprendido entre el periodo fetal (fundamental en la diferenciación de células totipotentes) y adolescencia marca un espacio de particular susceptibilidad a éstos. Una de las hipótesis más estudiadas es el fenotipo ahorrativo, en donde se postula que la inadecuada nutrición en etapas tempranas de la vida induce a resistencia a la insulina con el consiguiente riesgo de desarrollo de diabetes y nefropatía [37].
Múltiples estudios sobre epigenética y metabolismo se han llevado a cabo para conocer el inicio y la progresión de la enfermedad, estos apuntan a diversos factores y procesos biológicos que se desregulan activando procesos inflamatorios, vías metabólicas, estrés oxidativo y la remodelación de la función celular y su morfología [38].
Ejemplo de esto son estudios en humanos y en ratones en que se demuestra que el estrés perinatal afecta a la metilación del ácido desoxirribonucleico del receptor de glucocorticoides, proceso que acompaña al individuo durante toda su vida y regula la adiposidad corporal con el consiguiente aumento de riesgo de enfermedades cardiovasculares [39].
Múltiples vías llevan al deterioro de la función renal interviniendo en algún nivel en los mecanismos que perpetúan el daño microvascular. Idealmente el mejor mecanismo para evitar la progresión seria mantener un nivel glicémico adecuado, cuando esto no se logra, se desencadena una respuesta biológica patológica. Los productos avanzados de glicosilación determinan una cascada molecular en varios niveles que llevarán a la formación de productos biológicos dañinos a nivel microvascular.
El estrés oxidativo tiene un efecto deletéreo directo sobre las células mesangiales y membranas celulares endoteliales, por lo que controlar las especies reactivas del oxígeno también podría ayudar a disminuir la progresión de la enfermedad. Efecto similar tiene la vía de los polioles, en donde por el exceso de azúcar en sangre existe un desbalance del poder reductor, determinando un mayor estrés oxidativo. La vía de la proteína kinasa C también ejerce un efecto a nivel oxidativo, ya que determina la formación de especies reactivas del oxígeno. Sin embargo, esta vía no sólo produce daño por estrés oxidativo, también produce alteraciones a nivel mesangial, de membrana basal e intersticio renal. Todo ello altera la función renal.
El sistema renina angiotensina produce efectos deletéreos a distintos niveles, altera células y matriz extracelular, ya que la angiotensina II no sólo ejerce un efecto vascular, también tiene la capacidad de inducir la expresión de factores profibróticos que producen proliferación de matriz y llevan a fibrosis del glomérulo. Los factores de crecimiento que mencionamos se activan por el estado hiperglicémico y éstos son los responsables de la formación de tejido fibrótico y alteración de la vasculatura renal.
El componente inmune de la enfermedad también es importante, ya que las citoquinas inflamatorias son capaces de inducir una mayor respuesta inmune, alterar el estado hemodinámico a nivel intraglomerular, producir hipertrofia renal y, por si no fuera suficiente, también alteran la matriz extracelular de células mesangiales.
Con respecto al papel de la genética, principalmente está dado por la mayor susceptibilidad a producir ciertos factores de crecimiento y citoquinas inflamatorias, por lo que algunas personas tendrían una respuesta diferente de producción de estas sustancias biológicas.
Actualmente, los tratamientos contra esta patología están destinados a bloquear alguno de estos eslabones. Con ello se busca mejorar la filtración glomerular y disminuir la proteinuria. Dentro del pilar básico del manejo de la nefropatía diabética están el control glicémico y de tensión arterial estricto, así como el bloqueo del sistema renina angiotensina aldosterona. Respecto a este último punto, está demostrado el rol de los inhibidores de la enzima convertidora de angiotensina y bloqueadores del receptor de angiotensina II. Éstos disminuyen la progresión a enfermedad renal terminal, más por su efecto renoprotector que por su efecto hipotensor. No obstante, existe un 20% de casos que igualmente progresan, para lo cual se han diseñado diferentes fármacos que bloquean otras rutas fisiopatológicas tales como tirosina quinasa, fosfatasas y proteína kinasa dependiente de adenosina mono fosfato. Estos fármacos prometen a futuro, pero requieren estudios clínicos para validar sus resultados teóricos y así sopesar de mejor manera el costo/beneficio en los pacientes seleccionados para la intervención [40].
La nefropatía diabética es una importante complicación microvascular en la diabetes, ya sea por la comorbilidad que provoca en el paciente que la padece como por lo complejo de sus vías patogénicas, su interrelación y el daño renal que producen.
Si bien las vías de injuria están identificadas, el mejor entendimiento de éstas y el conocimiento genético agregado son de vital importancia para el desarrollo de terapias y de mecanismos paliativos para esta complicación vascular, en una enfermedad como la diabetes que tiene una alta prevalencia mundial.
Declaración de conflictos de intereses
Los autores han completado el formulario de declaración de conflictos intereses del ICMJE traducido al castellano por Medwave, y declaran no haber recibido financiamiento para la realización del reporte; no tener relaciones financieras con organizaciones que podrían tener intereses en el artículo publicado, en los últimos tres años; y no tener otras relaciones o actividades que podrían influir sobre el artículo publicado. Los formularios pueden ser solicitados contactando al autor responsable o a la dirección editorial de la Revista.
Financiamiento
Los autores declaran que no hubo fuentes de financiación externas.
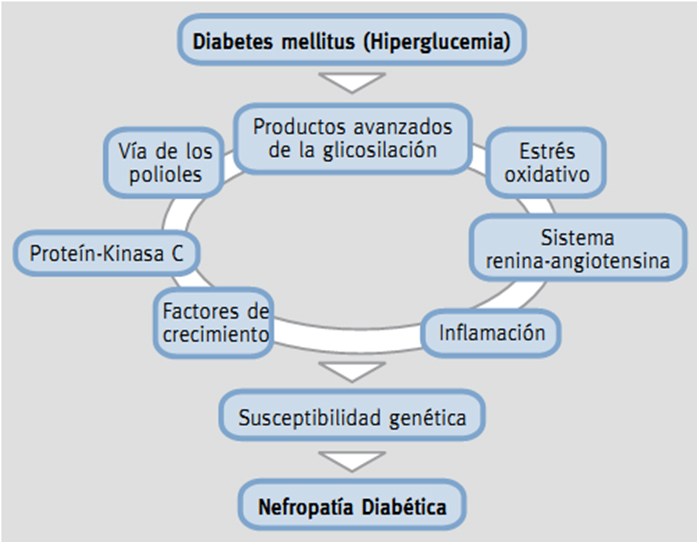 Figura 1. Factores que participan en el desarrollo de nefropatía diabética.
Figura 1. Factores que participan en el desarrollo de nefropatía diabética.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Chronic kidney disease is a common complication of diabetes. Its importance lies in its high prevalence and future projection. It is associated with high health costs and global cardiovascular deterioration as well. The development of this disease pathophysiology is being studied and it is known that a series of complex molecular pathways determining a microvascular disease are involved. This review addresses the known pathways in the development of diabetic nephropathy aiming to improve the understanding of potential therapeutic targets that could be developed in the future.
Autores:
Carlos Eduardo Meza Letelier[1], Camilo Alfredo San Martín Ojeda[1], José Javier Ruiz Provoste[1], Cristobal Jesus Frugone Zaror[1]
Citación: Meza Letelier CE, San Martín Ojeda CA, Ruiz Provoste JJ, Frugone Zaror CJ. Pathophysiology of diabetic nephropathy: a literature review. Medwave 2017 Ene-feb;16(1):6839 doi: 10.5867/medwave.2017.01.6839
Fecha de envío: 28/10/2016
Fecha de aceptación: 12/12/2016
Fecha de publicación: 12/1/2017
Origen: no solicitado
Tipo de revisión: con revisión por tres pares revisores externos, a doble ciego
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006 Nov;3(11):e442. | PubMed |Organizacion mundial de la salud. Definition, Diagnosis and Classification of Diabetes Mellitus and its Complications. Ginebra. WHO;1999. | Link |Gobierno de Chile Ministerio de Salud. [Acquired immunodeficiency syndrome]. Rev Chilena Infectol. 2010 Jun;27(3):239-76. | CrossRef | PubMed |Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio E, Ingelsson E, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010 Jun 26;375(9733):2215-22. | CrossRef | PubMed |Gobierno de Chile, Ministerio de Salud. Encuesta nacional de salud ENS Chile 2009-2010. Chile: Minsal, 2010 Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005 Jun;54(6):1615-25. | PubMed |Mount DB, Pollak MR, editors. Molecular and genetic basis of renal disease. Elsevier; 2008. | Link |Koeppen B, Stanton B. El sistema renal en: Fisiologia. Elsevier Sanders; 2009: 557-577. Schrijvers BF, De Vriese AS, Flyvbjerg A. From hyperglycemia to diabetic kidney disease: The role of metabolic, hemodynamic, intracellular factors and growth factors/cytokines. Endocr Rev. 2004 Dec;25(6):971-1010. | CrossRef | PubMed |Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol. 2003 May;14(5):1358-73. | PubMed |Cortes P, Méndez M, Riser BL, Guérin CJ, Rodríguez-Barbero A, Hassett C, et al. F-actin fiber distribution in glomerular cells: structural and functional implications. Kidney Int. 2000 Dec;58(6):2452-61. | PubMed |Fliser D, Wagner KK, Loos A, Tsikas D, Haller H. Chronic angiotensin II receptor blockade reduces (intra)renal vascular resistance in patients with type 2 diabetes. J Am Soc Nephrol. 2005 Apr;16(4):1135-40. | PubMed |Najafian B, Alpers CE, Fogo AB. Pathology of human diabetic nephropathy. Contrib Nephrol. 2011;170:36-47. | CrossRef | PubMed |Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, et al. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010 Apr;21(4):556-63. | CrossRef | PubMed |Mora-Fernandez C, Macías Heras M, Martínez-Castellao A, Gorriz Teruel J, De Alvaro Moreno F, Navarro-Gonzalez F. Fisiopatologia de la nefropatía diabética. Nefrología. 2008;1(1):28-38. | Link |Tan A, Forbes J, Cooper M. AGE, RAGE, and ROS in diabetic nephropathy. Seminars in nephrology. 2007 Apr 10;27(2):130–43. | CrossRef | PubMed |Hernández JC, Licea Puig ME, Hernándes García P, Abraham Marcel EA, Yanes Quesada M. Estrés oxidativo y diabetes mellitus. Rev Mex Patol Clin. 2011;58(1):4-15. | Link |Pawan Krishan VA. Diabetic nephropathy: aggressive involvement of oxidative stress. J Pharm Educ Res. 2011;2(1):35-41. Hodgkinson AD, Sřndergaard KL, Yang B, Cross DF, Millward BA, Demaine AG. Aldose reductase expression is induced by hyperglycemia in diabetic nephropathy. Kidney Int. 2001 Jul;60(1):211-8. | PubMed |Sung JK, Koh JH, Lee MY, Kim BH, Nam SM, Kim JH, et al. Aldose reductase inhibitor ameliorates renal vascular endothelial growth factor expression in streptozotocin-induced diabetic rats. Yonsei Med J. 2010 May;51(3):385-91. | CrossRef | PubMed |Xie P, Sun L, Oates PJ, Srivastava SK, Kanwar YS. Pathobiology of renal-specific oxidoreductase/myo-inositol oxygenase in diabetic nephropathy: its implications in tubulointerstitial fibrosis. Am J Physiol Renal Physiol. 2010 Jun;298(6):F1393-404. | CrossRef | PubMed |Inoguchi T, Sonta T, Tsubouchi H, Etoh T, Kakimoto M, Sonoda N, et al. Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J Am Soc Nephrol. 2003 Aug;14(8 Suppl 3):S227-32. | PubMed |Li J, Gobe G. Protein kinase C activation and its role in kidney disease. Nephrology (Carlton). 2006 Oct;11(5):428-34. | PubMed |Lee MR, Duan W, Tan SL. Protein kinase C isozymes as potential therapeutic targets in immune disorders. Expert Opin Ther Targets. 2008 May;12(5):535-52. | CrossRef | PubMed |Rodríguez-Iturbe B, Pons H, Herrera-Acosta J, Johnson RJ. Role of immunocompetent cells in nonimmune renal diseases. Kidney Int. 2001 May;59(5):1626-40. | CrossRef | PubMed |Yadav A, Vallabu S, Arora S, Tandon P, Slahan D, Teichberg S, et al. ANG II promotes autophagy in podocytes. Am J Physiol Cell Physiol. 2010 Aug;299(2):C488-96. | CrossRef | PubMed |Kumar V, Abbas AK, Aster JC. The endocrine pancreas En: Robbins & Cotran. Pathologic Basis of Disease. Elsevier Sanders; 2014: 1105-1117. Tuttle KR. Linking metabolism and immunology: diabetic nephropathy is an inflammatory disease. J Am Soc Nephrol. 2005 Jun;16(6):1537-8. | PubMed |Navarro JF, Mora C. Role of inflammation in diabetic complications. Nephrol Dial Transplant. 2005 Dec;20(12):2601-4. | PubMed |Navarro-González JF, Mora-Fernández C. The role of inflammatory Cytokines in diabetic Nephropathy. Journal of the American Society of Nephrology. 2008. Mar;19(3):433-42. | CrossRef | PubMed |Vilcek J. the cytokines: an overview, in the cytokine handbook, London, Elsevier Sanders, 2003:3-18. DiPetrillo K, Gesek FA. Pentoxifylline ameliorates renal tumor necrosis factor expression, sodium retention, and renal hypertrophy in diabetic rats. Am J Nephrol. 2004 May-Jun;24(3):352-9. | PubMed |Dai SM, Matsuno H, Nakamura H, Nishioka K, Yudoh K. Interleukin-18 enhances monocyte tumor necrosis factor alpha and interleukin-1beta production induced by direct contact with T lymphocytes: implications in rheumatoid arthritis. Arthritis Rheum. 2004 Feb;50(2):432-43. | PubMed |Navarro-González J, Mora-Fernández C, de Fuentes M, García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy: Abstract: Nature reviews Nephrology. Nature Reviews Nephrology. 2011 May 3;7(6):327–40. | CrossRef |Fermin I, Milagro Y. Epigenética en obesidad y diabetes tipo 2: papel de la nutrición, limitaciones y futuras aplicaciones. Rev Chil Endocrinol Diabetes. 2013;6(3):108-114. | Link |Conserva F, Gesualdo L, Papale M. A Systems Biology Overview on Human Diabetic Nephropathy: From Genetic Susceptibility to Post-Transcriptional and Post-Translational Modifications. J Diabetes Res. 2016;2016:7934504. | CrossRef | PubMed |






 Estudios originales
Estudios originales