Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Palabras clave:
Hígado graso, obesidad, estrés oxidativo, resistencia insulínica, ácidos grasos poli-insaturados de cadena larga de la serie n-3
Abreviaturas:
AGPI n-3: Ácidos Grasos Poli-Insaturados n-3 (ácido eicosapentaenoico [EPA] y ácido docosahexaenoico [DHS])
AGs: Ácidos Grasos
CAOX: Capacidad AntiOXidante del plasma
EHNA: EsteatoHepatitis No-Alcohólica
EROS: Especies Reactivas del Oxígeno
F2-I, F2-: isoprostanos productos de la peroxidación del ácido araquidónico
GSH: glutatión reducido
HGNA: Hígado Graso No-Alcohólico
HP: Hidroperóxidos Productos de la LP
LP: LipoPeroxidación, medida como producción de malondialdehido
NAFLD: Non-Alcoholic Fatty Liver Disease
O2•-: radical superóxido
POX: Proteínas OXidadas
PPAR-a(g): receptor activado por proliferadores peroxisomales-alfa(gama)
RI: Resistencia a la Insulina
SOD: SuperÓxido Dismutasa
SREBP-1c: proteína ligante al elemento regulador de esteroles-1c
TAGs: TriAcilGlicéridos
La enfermedad de Hígado Graso No-Alcohólico (HGNA) es la causa más importante de enfermedad hepática crónica y es considerada la manifestación hepática del síndrome metabólico asociado a obesidad y diabetes mellitus tipo 2. Se asocia a un amplio espectro de dańo hepático, que incluye tanto la esteatosis simple o acumulación intracelular de triacilglicéridos (TAGs), como la inflamación, fibrosis y cirrosis (esteatohepatitis no-alcohólica). Los mecanismos implicados son de carácter multifactorial, siendo la resistencia a la insulina un factor común que genera retención de ácidos grasos y TAGs dentro de los hepatocitos, con la producción de radicales libres a nivel mitocondrial capaces de inducir estrés oxidativo, producción de citoquinas y necrosis. Concomitantemente, se observa baja biodisponibilidad hepática de ácidos grasos poli-insaturados de cadena larga de la serie n-3, lo que alteraría la expresión de factores de transcripción asociados a la lipólisis y lipogénesis a nivel hepático. Un mejor conocimiento de los mecanismos etiopatogénicos del HGNA es fundamental para el desarrollo de estrategias terapéuticas eficaces a futuro.
En la actualidad, la enfermedad de Hígado Graso No-Alcohólico (HGNA) es la causa más importante de enfermedad hepática crónica relacionada al aumento en la incidencia de obesidad y diabetes mellitus tipo 2 en la población1. El HGNA se caracteriza por la presencia de esteatosis simple o acumulación intracelular de triacilglicéridos (TAGs), que puede progresar a la inflamación, fibrosis y cirrosis (EsteatoHepatitis No-Alcohólica, EHNA)2. El HGNA es considerado la manifestación hepática del síndrome metabólico, y su prevalencia en la población general alcanza el 15-20%, con una incidencia del 3% de EHNA. En sujetos con diabetes mellitus tipo 2 la incidencia es cercana al 50%, en la población obesa es de 76 a 90%, de los cuales alrededor del 35% desarrollará EHNA, mientras que la EHNA se presenta casi en la totalidad de los obesos mórbidos con diabetes2 3. La biopsia hepática es un factor clave para el diagnóstico del HGNA, que permite distinguir entre esteatosis simple, EHNA y grado de fibrosis. Sin embargo, existen técnicas como el ultrasonido, la tomografía computarizada o la resonancia magnética mediante las cuales se puede confirmar la presencia de esteatosis hepática con un alto grado de precisión2 4. Para su diagnóstico se requiere la exclusión del consumo de 20g/día de alcohol para las mujeres y 30g/día para los hombres, junto con descartar enfermedades virales, respuestas autoinmunes, factores hereditarios o metabólicos, drogas y toxinas5. El desarrollo de la resistencia a la insulina (RI) y del estrés oxidativo se consideran como los principales factores patogénicos del HGNA, los cuales, con la concurrencia de factores nutricionales, pueden determinar el inicio de la esteatosis y su progresión a la EHNA1 2 5. Las implicaciones patogénicas de estos factores son discutidos en este artículo de revisión, con el objeto de identificar mecanismos moleculares claves que pudiesen ser susceptibles de modificar por medio de intervenciones terapéuticas.
Alteraciones metabólicas, estrés oxidativo y resistencia a la insulina asociados a obesidad
Los mecanismos implicados en la acumulación de TAGs a nivel hepático y el subsiguiente daño hepatocelular son de carácter multifactorial y su naturaleza se está empezando a establecer. En condiciones normales, los ácidos grasos (AGs) son el principal combustible para el hígado. Sin embargo, en patologías como la obesidad, la gran afluencia de hidratos de carbono y lípidos induce cambios significativos en el metabolismo intermediario en el hígado.
Los elevados niveles de insulina (hiperinsulinemia) no son capaces de suprimir el flujo de AGs, mostrando un importante nivel de resistencia periférica a la acción de la insulina. El aumento en el pool de AGs circulantes es uno de los principales factores determinantes en la patogénesis del HGNA.
Es así como la hiperglicemia e hiperinsulinemia asociadas promueven la síntesis de AGs a partir de la glucosa e inhiben la β-oxidación de AG, siendo estos AGs re-direccionados hacia la formación de TAGs1 2 5. Considerando que la cantidad de AGs que son exportados vía lipoproteínas de muy baja densidad (VLDL) está limitada por la síntesis de su componentes proteicos, el exceso de AGs es principalmente convertido en TAGs y depositados en los hepatocitos con el consumo de dietas hipercalóricas. Debido a que el hígado tiene una capacidad limitada para la acumulación de TAGs, el depósito de lípidos en condiciones de sobrealimentación determina elevados niveles de AGs saturados, los que se asocian a disfunción y muerte celular. En efecto, estudios experimentales indican que el exceso de AGs condiciona altas tasas de b-oxidación, con producción de especies reactivas del oxígeno (EROS: radical superóxido [O2-] y peróxido de hidrógeno [H2O2]) a nivel de la cadena respiratoria mitocondrial, concomitantemente con la inducción de necrosis6. Estos resultados sugieren que la sobrealimentación puede inducir la sobrecarga de AGs en el hígado provocando altas velocidades de b-oxidación y formación de EROS, lo que está de acuerdo con los cambios en los parámetros relacionados con estrés oxidativo observados en el hígado de pacientes obesos con esteatosis7 10 (Figura 1). En efecto, en forma relativa a los valores controles, el hígado de los pacientes obesos presenta:
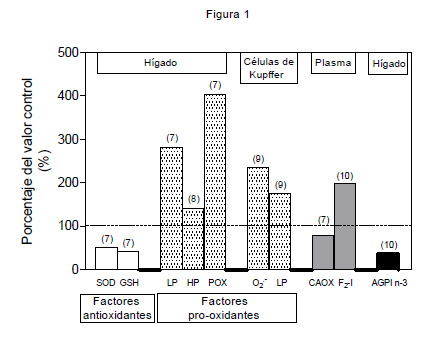 |
| Figura 1: Parámetros asociados al estrés oxidativo en pacientes obesos con hígado graso no-alcohólico, expresados como porcentaje de los valores controles. Los números entre paréntesis corresponden a las referencias específicas que contienen los datos. |
El desbalance redox observado en pacientes obesos representa un fenómeno de estrés oxidativo nutricional, que resulta de la ingesta excesiva y prolongada de combustibles metabólicos (carbohidratos y lípidos) y/o suministro inadecuado de antioxidantes dietarios11.
En condiciones de estrés oxidativo hepático, los pacientes obesos exhiben dos importantes alteraciones asociadas a este desbalance redox:
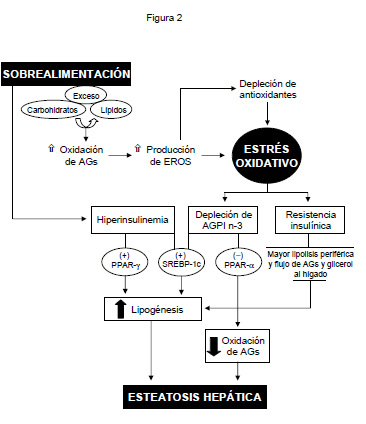 |
| Figura 2: Inducción de estrés oxidativo y su relación con la resistencia a la insulina (RI) y la esteatosis en el hígado graso no-alcohólico asociado a obesidad. |
Pérdida del control de la distribución metabólica de los ácidos grasos asociada a la obesidad
El estrés oxidativo asociado a obesidad tendría un papel causal en el desarrollo de la RI, condición en la cual la captación de glucosa por el músculo y tejido adiposo y la inhibición de la producción de glucosa por el hígado no responden adecuadamente a la insulina, mientras que los efectos pro-lipogénicos de la insulina son mantenidos a nivel hepático14. Este fenómeno se asocia a un cambio en el tipo de fosforilación del receptor de insulina y sus sustratos, que normalmente ocurre en residuos de tirosina, a residuos de serina, mediada por la activación de diversas serina-quinasas activadas por estrés oxidativo15. Esta modificación específica disminuye la fosforilación del receptor de insulina en tirosina y la vía de señalización intracelular de la hormona gatillando la RI12, alteración que podría ser facilitada por la depleción de AGPI n-3, por pérdida del grado de insaturación de los fosfolípidos de membrana que es requerido para el funcionamiento normal del receptor de insulina1.
Los AGPI n-3 ejercen sus efectos regulando el metabolismo de los lípidos en el hígado vía modificación en la transcripción génica, ya sea inhibiendo la expresión y procesamiento del factor pro-lipogénico proteína ligante al elemento regulador de esteroles-1c (SREBP-1c), y activando al receptor activado por proliferadotes peroxisomales-a (PPAR-a) que controla la expresión de los genes de enzimas de la oxidación de los AGs1 16. La disminución del contenido hepático de los AGPI n-3 en la obesidad se ha relacionado con:
Desde el punto de vista funcional, la depleción de los AGPI n-3 favorecería la esteatosis hepática al desactivar PPAR-a, lo cual disminuye la capacidad de oxidación de AGs, conjuntamente con incrementar la capacidad lipogénica del hígado al activar a SREBP-1c18 (Figura 2). De acuerdo con estos cambios, se ha observado que los pacientes obesos hiperinsulinémicos presentan mayor lipogenesis de novo19 20, proceso que estaría favorecido por la concomitante activación del factor pro-lipogénico PPAR-g21, además de SREBP-1c18, y la mayor lipólisis periférica asociada (Figura 2)1 22. Debido a la desactivación de PPAR-a relacionada con la depleción de los AGPI n-3 hepáticos en la obesidad, se produce un aumento significativo en la relación SREBP-1c/PPAR-a18, lo que determina un desbalance metabólico entre la lipogénesis y la oxidación de AGs a favor de la síntesis de AGs, lo que determinaría la esteatosis hepática (Figura 2)22.
El desarrollo de la esteatosis hepática en el paciente obeso es el resultado de múltiples alteraciones metabólicas que ocurren en condiciones de desbalance dietario y que involucran la inducción de estrés oxidativo, RI, junto con niveles alterados de ciertas adipoquinas capaces de influir en la sensibilidad a la insulina (Figura 2), y que puede progresar hacia la EHNA. Se ha sugerido que la cirugía bariátrica es el procedimiento más efectivo para lograr el control del peso corporal a largo plazo en la obesidad mórbida, la cual reduce la esteatosis, la RI y las alteraciones metabólicas asociadas22 23. Un adecuado tratamiento dietético para favorecer la pérdida de peso, como un aspecto terapéutico central, podría ser combinada con la administración de:
De acuerdo a estas consideraciones, estrategias terapéuticas que aumenten la sensibilidad a la insulina y las defensas antioxidantes en el hígado merecen ser evaluadas en futuros estudios controlados, con el fin de disminuir la esteatosis y evitar su progresión a la EHNA.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Non-alcoholic fatty liver disease (NAFLD) is the most important cause of chronic liver disease and is considered the hepatic manifestation of the metabolic syndrome associated with obesity and diabetes type 2. NAFLD refers to a wide spectrum of liver damage, including simple steatosis and inflammation, fibrosis, and cirrhosis (non-alcoholic steatohepatitis). The mechanisms involved in NAFLD are multifactorial, insulin resistance being a common factor in the retention of fatty acids and triacylglycerides within hepatocytes with mitochondrial production of free radicals, which trigger oxidative stress, cytokine production, and necrosis. Concomitantly, reduced availability of long chain n-3 polyunsaturated fatty acids is attained, a condition that alter the function of several transcription factors involved in lipolytic and lipogenic processes in the liver. A greater knowledge of the etiopathogenic mechanisms of NAFLD is crucial for the development of future effective therapeutic strategies.
Autores:
Luis Alberto Videla[1], Ana María Obregón[2], Paulina Pettinelli[3]
Citación: Videla LA, Obregón AM, Pettinelli P. Pathology of nonalcoholic fatty liver disease (NAFLD) associated with obesity: pathogenetic mechanisms. Medwave 2011 Jul;11(07):e5068 doi: 10.5867/medwave.2011.07.5068
Fecha de envío: 10/5/2011
Fecha de aceptación: 27/5/2011
Fecha de publicación: 1/7/2011
Origen: no solicitado
Tipo de revisión: con revisión externa por 2 revisores a doble ciego
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Videla LA, Rodrigo R, Araya J, Poniachik J. Insulin resistance and oxidative stress interdependency in non-alcoholic fatty liver disease. Trends Mol Med. 2006 Dec;12(12):555-8. Epub 2006 Oct 17. | CrossRef | PubMed |Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002 Apr 18;346(16):1221-31. | CrossRef | PubMed |Anderson N, Borlak J. Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol Rev. 2008 Sep;60(3):311-57. | CrossRef | PubMed |Saadeh S, Younossi ZM, Remer EM, Gramlich T, Ong JP, Hurley M, Mullen KD, Cooper JN, Sheridan MJ. The utility of radiological imaging in nonalcoholic fatty liver disease. Gastroenterology. 2002 Sep;123(3):745-50. | CrossRef | PubMed |Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006 Feb;43(2 Suppl 1):S99-S112. | CrossRef | PubMed |Aronis A, Madar Z, Tirosh O. Mechanism underlying oxidative stress-mediated lipotoxicity: exposure of J774.2 macrophages to triacylglycerols facilitates mitochondrial reactive oxygen species production and cellular necrosis. Free Radic Biol Med. 2005 May 1;38(9):1221-30. | CrossRef | PubMed |Videla LA, Rodrigo R, Orellana M, Fernandez V, Tapia G, Quińones L, Varela N, Contreras J, Lazarte R, Csendes A, Rojas J, Maluenda F, Burdiles P, Diaz JC, Smok G, Thielemann L, Poniachik J. Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin Sci (Lond). 2004 Mar;106(3):261-8. | CrossRef | PubMed |Oliveira CP, Faintuch J, Rascovski A, Furuya CK Jr, Bastos Mdo S, Matsuda M, Della Nina BI, Yahnosi K, Abdala DS, Vezozzo DC, Alves VA, Zilberstein B, Garrido AB Jr, Halpern A, Carrilho FJ, Gama-Rodrigues JJ. Lipid peroxidation in bariatric candidates with nonalcoholic fatty liver disease (NAFLD) -- preliminary findings. Obes Surg. 2005 Apr;15(4):502-5. | CrossRef | PubMed |Malaguarnera L, Di Rosa M, Zambito AM, dell'Ombra N, Nicoletti F, Malaguarnera M. Chitotriosidase gene expression in Kupffer cells from patients with non-alcoholic fatty liver disease. Gut. 2006 Sep;55(9):1313-20. Epub 2006 Jul 6. | CrossRef | PubMed | PMC |Elizondo A, Araya J, Rodrigo R, Signorini C, Sgherri C, Comporti M, Poniachik J, Videla LA. Effects of weight loss on liver and erythrocyte polyunsaturated fatty acid pattern and oxidative stress status in obese patients with non-alcoholic fatty liver disease. Biol Res. 2008;41(1):59-68. Epub 2008 Aug 21. | CrossRef | PubMed |Sies H, Stahl W, Sevanian A. Nutritional, dietary and postprandial oxidative stress. J Nutr. 2005 May;135(5):969-72. | PubMed |Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006 Apr 13;440(7086):944-8. | CrossRef | PubMed |Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond). 2004 Jun;106(6):635-43. | CrossRef | PubMed |George J, Liddle C. Nonalcoholic fatty liver disease: pathogenesis and potential for nuclear receptors as therapeutic targets. Mol Pharm. 2008 Jan-Feb;5(1):49-59. Epub 2007 Dec 12. | CrossRef | PubMed |Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. 2005 Jul-Aug;7(7-8):1040-52. | CrossRef | PubMed |Clarke SD. Nonalcoholic steatosis and steatohepatitis. I. Molecular mechanism for polyunsaturated fatty acid regulation of gene transcription. Am J Physiol Gastrointest Liver Physiol. 2001 Oct;281(4):G865-9. | PubMed |Araya J, Rodrigo R, Pettinelli P, Araya AV, Poniachik J, Videla LA. Decreased liver fatty acid delta-6 and delta-5 desaturase activity in obese patients. Obesity (Silver Spring). 2010 Jul;18(7):1460-3. Epub 2009 Oct 29. | CrossRef | PubMed |Pettinelli P, Del Pozo T, Araya J, Rodrigo R, Araya AV, Smok G, Csendes A, Gutierrez L, Rojas J, Korn O, Maluenda F, Diaz JC, Rencoret G, Braghetto I, Castillo J, Poniachik J, Videla LA. Enhancement in liver SREBP-1c/PPAR-alpha ratio and steatosis in obese patients: correlations with insulin resistance and n-3 long-chain polyunsaturated fatty acid depletion. Biochim Biophys Acta. 2009 Nov;1792(11):1080-6. Epub 2009 Sep 4. | PubMed |Schwarz JM, Linfoot P, Dare D, Aghajanian K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr. 2003 Jan;77(1):43-50. | PubMed |Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005 May;115(5):1343-51. | PubMed | PMC |Pettinelli P, Videla LA. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab. 2011 May;96(5):1424-30. Epub 2011 Feb 16. | CrossRef | PubMed |Videla LA. Oxidative stress and insulin resistance as interdependent pathogenic mechanisms in non-alcoholic fatty liver disease associated with obesity. En: Free Radical Pathophysiology, Alvarez S, Evelson P, eds. Transworld Research Network; Kerala, India 2008:369-85. Angulo P. NAFLD, obesity, and bariatric surgery. Gastroenterology. 2006 May;130(6):1848-52. | CrossRef | PubMed |Capanni M, Calella F, Biagini MR, Genise S, Raimondi L, Bedogni G, Svegliati-Baroni G, Sofi F, Milani S, Abbate R, Surrenti C, Casini A. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: a pilot study. Aliment Pharmacol Ther. 2006 Apr 15;23(8):1143-51. | CrossRef | PubMed |






 Estudios originales
Estudios originales