Revista Biomédica Revisada Por Pares

Este texto completo es la transcripción editada y revisada de una conferencia dictada en el marco de las reuniones clínicas del Servicio de Pediatría del Complejo de Salud San Borja-Arriarán. La publicación de estas actas científicas ha sido posible gracias a una colaboración editorial entre Medwave y el Servicio de Pediatría. El jefe de Servicio es el Dr. Francisco Barrera y el coordinador de las Reuniones Clínicas es el Dr. Luis Delpiano.
El paciente corresponde a un recién nacido (RN), de sexo masculino, nacido a las 40 semanas de gestación que fue adecuado para su edad gestacional, segundo hijo de un embarazo controlado que nació por cesárea debido a un síndrome hipertensivo del embarazo en la ciudad de Coyhaique. No existen antecedentes de hallazgo antenatal ecográfico relacionado con esta patología. El peso del recién nacido fue de 2,97 kilogramos, talla: 48,5 centímetros, Apgar 9-10.
Al nacer el RN se encontraba en buenas condiciones generales, pero al segundo día de vida presentó dificultad respiratoria y cianosis, dificultad para alimentarse, evolucionando progresivamente con requerimientos de oxígeno aumentados. En este contexto se solicitó una radiografía de tórax que mostraba una dextrocardia, cardiomegalia y congestión pulmonar. Se procedió a la realización de un ecocardiograma, que mostró un ductus arterioso persistente, situación por la que se inició tratamiento con prostaglandinas, obteniéndose una buena respuesta clínica inicial.
A pesar de lo anterior el RN persistió con dificultad respiratoria y requerimientos de oxígeno aumentados. En este contexto, se sospechó una cardiopatía congénita cianótica y el paciente fue trasladado al Hospital Luis Calvo Mackenna en la ciudad de Santiago, donde nuevamente se realizó un ecocardiograma que mostró la dextrocardia, un ductus arterioso que ya se encontraba cerrado, una estenosis relativa de la arteria pulmonar derecha y un foramen oval permeable, descartándose finalmente una cardiopatía congénita que explicara el cuadro clínico en general.
A los 6 días el RN mantenía signología respiratoria obstructiva e hipersecreción bronquial, además de pesquisarse una conjuntivitis aguda. Se solicitó una nueva radiografía que mostró un infiltrado intersticial bilateral (Fig.1). Además se realizó una inmunofluorescencia y PCR (reacción en cadena de polimerasa), por sospecha de algún cuadro infeccioso como Chlamydia trachomatis y ureaplasma que resultaron negativos. Se tomó un cultivo de la secreción conjuntival cuyo resultado fue positivo para Haemophilus influenzae sp., que fue tratado con cloranfenicol. A pesar de todo el RN persistía con requerimientos de oxígeno frente a una FiO2 de 0,5.
A los 17 días de vida se trasladó al departamento de Pediatría del Hospital Clínico San Borja Arriarán. El peso al ingreso fue de 3,850 kg, presentando requerimientos de oxígeno más bajos: 0,25 litros por naricera. Además presentaba congestión nasal, signología obstructiva, e hipersecreción bronquial. Fue evaluado por médicos broncopulmonares y también por otorrinolaringólogos, quienes, en el contexto de una dextrocardia, signología respiratoria y congestión pulmonar, plantearon la sospecha de un síndrome de Kartagener. Se decidió diferir el estudio mediante biopsia de carina para el ańo de vida.
Se procedió a realizar una ecografía abdominal que mostró una imagen en espejo de todos los órganos intraabdominales. Un nuevo ecocardiograma mostró dextrocardia, foramen oval permeable y estenosis fisiológica de rama pulmonar derecha. Finalmente fue traslado a Coyhaique al mes de vida con requerimientos de O2 0,3 L/min en forma permanente.
Los diagnósticos de egreso fueron: situs inversus total, observación síndrome de Kartagener, oxigenodependencia, foramen oval permeable, y estenosis relativa de la arteria pulmonar derecha.
En general, se podría decir que los seres humanos son externamente simétricos, pero internamente asimétricos. Se dispone de una descripción histórica de autopsia correspondiente al Virrey de México, el Marqués de las Amarillas, realizada muchos ańos después de su fallecimiento. El escrito de su autopsia dice así: “Hecha patente según las reglas del arte la cavidad del pecho, se hizo visible el corazón sin pericardio inclinada su punta y cuerpo al lado derecho, y su base miraba al lado izquierdo… La arteria pulmonar salía del ventrículo derecho del corazón colocada a el lado izquierdo, se deslizaba oblicuamente hacia el lado derecho, lo que me hizo creer, que los pulmones habían mudado también de situación: en efecto el derecho estaba dividido en dos lobos, y el izquierdo en tres”. Lo anterior fue descrito por Domingo Russi en el ańo 1760.
Siempre se había seńalado que Mathew Baillie, en 1788, había sido el primero en describir esta condición, pero ahora se sabe que no fue así. En el ańo 1933 Kartagener reportó cuatro pacientes con la tríada de: sinusitis, bronquiectasias y situs inversus. Hacia el ańo 1976, Afzelius describió en hombres infértiles la presencia de esta misma tríada, planteándose básicamente que existiría un problema de cilios, por lo tanto los espermios estarían afectados también respecto de su movilidad.
A continuación se abordará la interrogante respecto de si los cilios jugarían o no, un rol en determinar lateralidad.
Las alteraciones de la lateralidad incluyen cambios tanto en orientación como en disposición de los órganos, y se clasifican en situs inversus totalis y situs ambiguus. El situs inversus totalis corresponde a la imagen en espejo del situs solitus, correspondiente a lo normal, se presenta en uno a seis mil u ocho mil RN y en él todos los órganos mantienen una posición con respecto a los otros órganos. El situs ambiguus corresponde a al menos un órgano impar con orientación contraria, presentándose en uno en diez mil RN.
Los seres humanos poseen algunos órganos que son pares, pero que a su vez son asimétricos entre sí, como por ejemplo: los pulmones, donde el pulmón derecho tiene tres lóbulos y el izquierdo tiene sólo dos lóbulos; eso corresponde a la asimetría normal.
El isomerismo corresponde a la alteración en la asimetría de órganos pares, generalmente bronquios, pulmones y aurículas. Se puede clasificar en isomerismo izquierdo y derecho.
El isomerismo izquierdo corresponde a órganos que tienen una manifestación de predominio izquierdo, como la presencia de dos aurículas largas y estrechas, dos pulmones bilobulados, bronquios por debajo de la arteria pulmonar, asociados a poliesplenia. Por su parte, el isomerismo derecho corresponde a órganos con una manifestación predominante a derecha, como aurículas en forma de pirámide, dos pulmones trilobulados, bronquios sobre arteria pulmonar, asociados a asplenia; también se le conoce como síndrome de Ivemark.
De todos modos estas clasificaciones no son útiles para el manejo de los pacientes, porque lo importante en ese sentido es tener una descripción detallada de la disposición y la morfología de los órganos.
En la Fig. 2 se observa el situs solitus, situs inversus, isomerismo derecho (dos pulmones trilobulados y la ausencia de bazo) y, por último, el isomerismo izquierdo donde los pulmones son bilobulados con poliesplenia.
En la Fig. 3 se aprecian imágenes de radiografías de tórax correspondientes a diversas alteraciones de la lateralidad.
El situs ambiguus se asocia mucho más a cardiopatías que los situs inversus totales o que un situs solitus. En 80% de los casos, el situs ambiguus se asocia con malformaciones cardiacas tales como transposición de grandes vasos (TGA), doble salida de ventrículo derecho, doble entrada de ventrículo izquierdo, comunicación interauricular (CIA), comunicación interventricular (CIV), retorno venoso anómalo y aurículas isoméricas. También se presentan más anomalías gastrointestinales tales como estómago en posición derecha, alteraciones de la vía biliar, hígado en la línea media, malrrotación intestinal, anesplenia y poliesplenia.
El situs inversus tiene mucho menos frecuencia de cardiopatías asociadas que el ambiguo, sólo entre 3 y 6%. Se desconoce el motivo, pero es posible que durante la formación de los órganos la disposición de éstos presentes pueda ser un factor determinante. En el caso de un situs ambiguus podría existir mayor riesgo de asociación con cardiopatías por la disposición que adoptan los órganos. Por otro lado, el situs solitus se relaciona con cardiopatías sólo en un 0,6%.
Dentro de los mecanismos que determinan lateralidad izquierda-derecha, podemos encontrar:
Todos estos datos son extraídos de estudios en embriones, realizados en ratones, peces cebra y ranas, que tratan de explicar las alteraciones de las asimetrías.
La seńal más temprana ocurre a nivel del nodo de Hensen, que corresponde a un conjunto de células que se forman en el extremo cefálico de la línea primitiva, durante la etapa embrionaria de gastrulación, cuando el embrión tiene dos capas germinales correspondientes a mesodermo y endodermo (Fig. 4). En esta etapa el embrión es totalmente simétrico, pero existiría alguna seńal, que todavía no es bien conocida, que determinaría la lateralidad, o sea el que algunas cosas se vayan hacia el lado izquierdo y otras hacia el derecho. Entonces habría, en un comienzo, una alteración en las células del nódulo, en cuanto a su polaridad, que determinarían el irse hacia un lado y no hacia el otro.
Figura 4. Esquema embrionario donde se muestra el nodo de Hensen y línea primitiva.
Respecto de las seńales que participan en el nodo, en cuanto a estructura y función ciliar, en general existe una coordinación en la formación del eje izquierda-derecha en todos los vertebrados, pero las estructuras que determinan esto son diferentes en cada tipo de ellos. La formación del eje izquierda-derecha comienza muy temprano, antes de la organogénesis. Además, se ha visto que la formación del nodo en ratones es a los 7,5 a 8,5 días, mientras en humanos esto ocurre entre los 7 y 10 días. En consecuencia, se trata de una etapa muy temprana de la fecundación donde ya empieza a haber diferenciación.
Los modelos animales muestran en el “nodo” la presencia de células que tienen cilios, siendo éstos móviles y capaces de generar un “flujo nodal”, el que a su vez explicaría de cierta forma la transferencia de ciertos genes, hacia un lado y no hacia el otro. Los cilios realizarían un movimiento especial, de carácter rotacional, que llevaría este flujo hacia un lado.
En modelos animales, se ha visto que hay dos modelos de cilios: centrales y periféricos, siendo los centrales móviles y los periféricos inmóviles, que serían los encargados de alguna manera de la parte sensitiva.
Los cilios centrales están conformados normalmente por nueve pares de microtúbulos periféricos y dos microtúbulos individuales centrales, lo que se conoce como conformación “9+2” (Fig. 5).
Figura 5. Esquema que ilustra modelo de cilio conformado por microtúbulos “9+2”.
En etapas tempranas de la embriogénesis los cilios tienen una conformación “9+0”. Llama la atención que el no poseer este par de microtúbulos de igual forma permite a los cilios centrales de los embriones tener movilidad, determinando así la lateralidad. Las células del nodo con cilios móviles tienen proteínas de dineína izquierda-derecha, que de cierta forma otorgan la factibilidad para que los cilios se muevan y se deben a la expresión del gen DNAH11, que está relacionado también con algunos tipos de disquinesia ciliar primaria.
Dentro de las seńales en el nodo que participan en la estructura y función ciliar se encuentra el factor Foxj1 (Fig. 6), que participaría en la regulación de la motilidad de éstos. En los cilios además existe un transporte intraflagelar, responsable de movilizar las proteínas hacia las zonas apical o basal, otorgando de este modo funcionalidad al cilio.
Figura 6. Seńales en el nodo que participan en la estructura y función ciliar.
Factores genéticos
Previamente se mencionó la existencia de un flujo nodal hacia izquierda que llevaría a la expresión simétrica de los genes. En la Fig. 7 se esquematiza el control molecular de la asimetría en un modelo de pollo. En la zona inferior de ésta, se observa el nodo de Hensen donde se produciría un factor de transcripción Shh que estimularía a los factores Car y Nodal, que a su vez estimulan a un segundo factor Nodal y después al Pitx2,correspondiente a un gen que está relacionado con la organogénesis del corazón, pulmones e intestino. En el lado contrario de la figura se observa que el factor shh está bloqueado. En primera instancia la activina beta B estimula el Bmp4, quién es el encargado de inhibir al Shh, bloqueándolo, provocando que las cascadas que se dan al lado izquierdo y el derecho sean diferentes. Cada lado tendría vías de seńalización específicas.
Figura 7. Cascadas de mediadores relacionados a la asimetría.
El patrón de expresión nodal se encuentra muy conservado en todas las especies estudiadas, desde el pez cebra hasta el hombre. El factor Nodal pertenece a la familia del factor de crecimiento transformante beta (TGF-B), y dentro de sus características están manifestarse transitoriamente en el mesodermo lateral izquierdo y estar directamente relacionado con lateralidad del corazón y otros órganos. Induce la expresión de gen Pitx 2 que continúa actuando mientras los esbozos viscerales realizan su morfogénesis, además activa los genes antagonistas Lefty-1 y Lefty-2 a través de factores de crecimiento, que se relacionan también con la diferenciación del lado izquierdo.
El gen Pixt 2 está relacionado con la organogénesis de corazón, intestino y pulmón, siendo responsable en gran medida de la expresión asimétrica.
En órganos como el corazón, los mecanismos que específicamente contribuyen a que la aurícula derecha sea diferente de la izquierda aún no son bien conocidos.
Existen muchos genes involucrados en las asimetrías (Fig. 8). La mayoría de ellos posee herencia autosómica recesiva, aunque el gen Zic 3 presenta herencia ligada a los cromosomas X e Y, relacionándose a heterotaxia mediante la proteína Zic 3. Por otro lado, el gen ACVR 2B, mediante la activina, se relaciona con situs ambiguus.
Figura 8. Genes relacionados a las asimetrías (11).
Factores ambientales
Existen factores ambientales que también podrían explicar las alteraciones de la simetría.
La diabetes tipo 1 en el embarazo se asocia con mayor frecuencia a alteraciones del isomerismo de las aurículas. Asimismo, ratones diabéticos no obesos con hiperglicemia precoz han manifestado alteraciones de la lateralidad, presentando también una mayor frecuencia situs ambiguus. Otros factores serían el ácido retinoico y derivados de la vitamina A.
żFamiliar o esporádico?
Las alteraciones de la simetría se presentan de forma familiar en menos de un 5% de los casos, relacionándose con un gen de herencia autosómica dominante, responsable de la heterotaxia, pero que se expresa escasamente en la población general, probablemente debido a que se asocia a una menor transmisibilidad del alelo mutado.
La heterotaxia puede encontrarse de forma aislada o asociada a algún síndrome. Se han descrito más de cincuenta y nueve síndromes asociados a heterotaxia. Entre ellos están: el síndrome de Kartagener; el síndrome de Bardet-Bield, caracterizado por retardo mental, obesidad, polidactilia, retinitis pigmentosa, e insuficiencia renal, relacionados al gen ttc 8; la enfermedad renal quística; anomalías genitourinarias y ano imperforado; además del síndrome de Poland que en algunas ocasiones está asociado a dextrocardia.
La DCP presenta una alteración específica a nivel ciliar, comprometiendo la motilidad a nivel estructural, provocando una disfunción del clearance mucociliar, barrido ciliar cuyas funciones son eliminar los gérmenes, detritus y otros.
La DCP posee una prevalencia que varía en distintas series entre 1:40.000 (Torgensen), 1:22.000 – 1:4.100 (Japón) Afzelius, siendo este último un valor obtenido en una muestra de pacientes que habían sobrevivido a la exposición a la bomba atómica en Japón durante la segunda guerra mundial. Se ha descrito también que un 13% de las bronquiectasias difusas corresponden a pacientes con disquinesia ciliar primaria. Las bronquiectasias, dentro de la DCP, se presentan tardíamente debido a que el proceso de alteración del clearance ciliar no ocurre en forma inmediata, por lo tanto la radiografía no mostraría alteración alguna en un comienzo. La mediana de edad para el diagnóstico es entre 4,4 y 6 ańos, siendo generalmente más tardío es en aquellos pacientes que no tienen alteración de la lateralidad.
En la Fig. 9 se aprecia un cilio normal por microscopia electrónica, con sus nueve pares de microtúbulos periféricos y dos aislados centrales “9+2”, esquematizándose los brazos de dineína, internos y externos, encargados de la movilidad. La conformación “9+0” es inmóvil, salvo en el embrión, donde los cilios de conformación “9+0” son móviles cuando se ubican centralmente.
Figura 9. Microscopía electrónica de cilio “9+2” (15).
Los defectos genéticos en la dineína pueden ocurrir en sus brazos externos, internos, en ambos o a nivel central de ésta. Entre 10 y 20% de los casos la microscopia electrónica no muestra defectos estructurales (16).
En la Fig. 10 se aprecian diversas imágenes que muestran las alteraciones estructurales de los cilios, evaluados a través de microscopía electrónica. Lo más frecuente es que las alteraciones en los brazos de dineína provoquen un cilio inmóvil; en otras oportunidades el cilio presenta un patrón de movimiento anormal, lo que también produce efectos en la simetría.
Los genes DNA11, DNAH11 y DNAH5, están asociados con DCP y síndrome de Kartagener. También hay otros genes que están involucrados a nivel del efector de centrómeros y la mayoría de éstos se heredan en forma autosómica recesiva.
Las condiciones que permiten sospechar la disquinesia ciliar primaria son, en el caso del período antenatal, ecografía que muestra alteraciones de lateralidad, como Situs inversus (25% DCP) y Situs ambiguus (6%). En el período neonatal, una rinitis persistente o un síndrome de distrés respiratorio del recién nacido podrían corresponder a signos de sospecha. En nińos las manifestaciones incluyen tos crónica, neumonía recurrente, SBO recurrente, rinitis crónica, otitis media recurrente, otorrea, hipoacusia, antecedentes de historia neonatal de distrés respiratorio, al igual que de asma en la familia, asma con deficiente respuesta a tratamiento habitual, hipoacusia o uso de colleras en la familia. En adultos, puede haber signología como tos persistente, bronquiectasias, tratamiento por asma atípica, poliposis nasal, rinosinusitis crónica, inmotilidad de los espermios en las personas adultas e historia de infertilidad, embarazo ectópico en mujeres.
Dentro de las principales patologías asociadas a DCP se encuentran las siguientes: cardiopatías congénitas, atresia biliar primaria, retinitis pigmentosa, hidrocéfalo, enfermedad renal y hepática poliquística, atresia esofágica y reflujo gastroesofágico severo. También es posible asociar DCP a otras ciliopatías, como nefronoptisis, síndrome Bardet- Bield, síndrome Alstrom y síndrome Meckel- Gruber.
Estudios demuestran que existe una disminución del flujo espiratorio forzado en el primer segundo (VEF1) en prácticamente todos los pacientes que poseen DCP, predominantemente acentuado en adultos respecto de nińos, radicando en este hecho la importancia de un diagnóstico certero lo más precozmente posible. Los principales síntomas en las series de adultos y nińos son la tos en el 100% de los casos, bronquiectasias en un 98% y 61% de pacientes adultos y nińos, respectivamente, sinusitis en un 47% y 45% de pacientes adultos y nińos, respectivamente, otitis media 92% y 100% en adultos y nińos respectivamente, historia de distrés respiratorio en la infancia en un 65% y 87% en adultos y nińos, respectivamente, y el situs inversus en un 46% y 68% en adultos y nińos, respectivamente. El VEF1 tiene una disminución de un 0,8% por ańo de vida en las DCP.
Otro aspecto importante en el diagnóstico precoz de las DCP, tiene relación con las infecciones, pues éstas son mucho más frecuentes en éstos pacientes respecto de la población normal. Resulta fundamental definir mediante cultivos y antibiogramas el tratamiento más adecuado. Los gérmenes más frecuentes aislados en nińos son Haemophilus influenzae, Pseudomonas, y Staphylococcus aureus.
Existen métodos de screening y otros de diagnóstico, propiamente tales. Dentro de los test de screening estaría la medición de la reducción de los niveles de óxido nítrico, el que se produce en el epitelio respiratorio a partir de L-arginina (Fig. 11) mediante la estimulación de cascadas inflamatorias. En la DCP no está estimulada la enzima óxido nítrico sintasa, por lo que su disfunción conlleva a niveles finales de óxido nítrico bajos.
Una dificultad con este test es que cualquier situación que provoque una obstrucción nasal también provocará que disminuya el óxido nítrico, como por ejemplo la poliposis. Otra dificultad tiene que ver con que el paciente necesita tener por lo menos entre 4 y 6 semanas sin infección para que no se altere falsamente el resultado, y que sus valores de referencia están determinados para pacientes entre 4 y 17 ańos o más, no para nińos menores por lo que en ellos se dificulta el diagnóstico.
Otro test de screening corresponde al test de sacarina, que consiste en administrar este elemento en los cornetes nasales inferiores del paciente, midiendo el tiempo en que éste comienza a sentir el gusto al producto químico. Actualmente este procedimiento está en desuso.
También como test de screening está el clearance mucociliar, que consiste en administrar al paciente mediante un aerosol, albúmina marcada con tecnecio, evaluando el tiempo que demora éste en limpiarse. Este método posee una sensibilidad de 88% y una especificidad de 100%, para nińos mayores de cinco ańos de edad.
En cuanto a diagnóstico tenemos el cepillado nasal mediante un instrumento llamado broncoscopio nasal, siendo un procedimiento rápido, pero que en nińos necesita sedación y ausencia de infección en las 4-6 semanas previas al procedimiento.
Un segundo método diagnóstico consiste en evaluar el movimiento de los cilios, pues si bien en algunas microscopías no hay una alteración estructural, sí puede haber un problema en el batido de los cilios. El procedimiento se realiza mediante el uso de una cámara de video que permite medir la frecuencia del movimiento de los cilios y las características de estos movimientos.
Un tercer método diagnóstico consiste en la evaluación de la estructura ciliar, mediante microscopía electrónica.
Un cuarto método corresponde al cultivo celular. Su principal ventaja consiste en disminuir de modo importante la presencia de falsos positivos y su desventaja es el necesitar un tiempo prolongado para la obtención de resultados.
Adicionalmente, están comenzando a emplearse técnicas de análisis de dineína por inmunofluorescencia, cuyo rol aún es potencial. También existe el análisis genético, que no está recomendado en un inicio, sino en segunda instancia para realizar consejería.
Un estudio diseńado para evaluar la sensibilidad y especificidad de los distintos exámenes, realizado a 270 pacientes con DCP que tenían microscopía electrónica negativa y 70 con DCP más microscopía electrónica positiva, concluyó que una frecuencia de batido ciliar de menos de 11 Hertz era altamente sensible y específica. La medición del patrón de movimiento ciliar en un score de 0 a 3, siendo 0 la presencia de movimientos normales y 3 muy alterado, arroja que el score con más de 2 puntos promedia una sensibilidad de un 92% y una especificidad de un 97%. El índice de inmotilidad, correspondiente a que los cilios no se muevan en más de un 10%, demostró tener una sensibilidad de un 69% y una especificidad de un 98%. Al combinar la frecuencia y patrón de movimiento o frecuencia y alteraciones de los patrones ciliares no cambiaron mucho las sensibilidades ni las especificidades ya descritas.
El tratamiento de la DCP debe ser multidisciplinario, incluyendo kinesioterapia respiratoria, ejercicio y evitar la exposición al humo del tabaco. Los cultivos frecuentes de esputo o secreciones inducidas mediante broncoscopía son importantes en pacientes en los que falla el tratamiento antibiótico usual o presentan atelectasias persistentes. De mucha importancia resulta también el uso de antibioterapia precoz, idealmente siempre teniendo gérmenes aislados.
La evaluación de función pulmonar resulta muy útil. Las radiografías de tórax, a pesar de su baja sensibilidad para detectar bronquiectasias en nińos pequeńos, sí son muy útiles en adultos y exacerbaciones. La realización de tomografía axial computada de tórax cada 4 o 5 ańos es recomendable, pues es mucho más específica para detectar malformaciones y bronquiectasias. Por último, la cirugía resulta ser totalmente excepcional, pudiendo realizarse lobectomías o segmentectomías en bronquiectasias o infecciones que resultan refractarias e irreversibles. También se puede recurrir a colleras en nińos que presentan otitis media recurrente.
Respecto de los antibióticos, éstos se utilizan según germen aislado. Ante la presencia de gérmenes multirresistentes, como Pseudomonas multirresistentes o mycobacterium no tuberculoso, se puede utilizar un esquema asociado de antibióticos endovenosos e inhalatorios, similar a lo indicado a ciertos pacientes con fibrosis quística. No hay evidencia suficiente para recomendar profilaxis antibiótica, y sólo debería considerarse en casos de los pacientes que han tenido infecciones y tratamientos antibióticos recurrentes.
Respecto de los fármacos inhalados, como parte del tratamiento de la DCP, tanto los agonistas de los receptores beta-2 adrenérgicos, la dornasa alfa (una desoxirribonucleasa humana recombinante “DNasa o ADNasa”) y las soluciones hipertónicas, no hay ensayos clínicos controlados ni aleatorizados que avalen su uso. La mayoría de los estudios de DNasa y soluciones hipertónicas son en pacientes con fibrosis quística.
Un estudio realizado en 19 nińos con DCP, doble ciego, de distribución aleatoria y cruzado, realizado para evaluar la eficacia del salbutamol en el tratamiento de su condición de salud, demostró que al aplicar salbutamol y luego placebo, no hubo diferencias estadísticamente significativas, por lo que se concluyó que el salbutamol no tendría una relevante utilidad como tratamiento de la disquinesia ciliar primaria.
Del mismo modo tampoco se posee evidencia suficiente para recomendar el uso de corticoides ni azitromicina como antiinflamatorio. Sólo se tienen datos por extrapolación de estudios en pacientes con fibrosis quística. Respecto de las kinesioterapias respiratorias, no existe evidencia que demuestre la superioridad de una técnica respecto de otras, dependiendo más bien de características del propio paciente, como edad, y se utiliza tanto como terapia de mantención, como en exacerbaciones. En relación a las inmunizaciones se recomienda la indicación de la antiinfluenza anual, antineumocócica y el uso de palivizumab cuando el diagnóstico es muy temprano, al mes de vida.
En el Hospital Roberto del Río, se publicó una experiencia de 38 pacientes, que presentaban una edad promedio de 10 ańos, siendo 18 mujeres y 20 hombres, teniendo además una edad de derivación promedio de 5 ańos. Un 84% de ellos había presentado síntomas antes del primer ańo de vida, y de ellos la mitad en el período de recién nacido. De todos los pacientes el 92% había presentado síndrome bronquial obstructivo (SBO), neumonía recurrente en un 73%, sinusitis en un 63% y otitis media recurrente en un 3,2%.
La edad de diagnóstico en promedio fue de 6 ańos, 6 pacientes presentaban situs inversus y 19 pacientes atelectasias crónicas. A 27 pacientes se les realizó TAC de tórax, obteniéndose como resultado que 8 de ellos resultaron normales y el resto presentaron atelectasias crónicas o atelectasias agudas en lóbulo medio o basal.
A 26 pacientes que se les realizó estudio de función pulmonar, obteniéndose como resultado que 7 de ellos presentaban limitación ventilatoria obstructiva leve. Cirugía solamente se realizó en 6 casos, correspondiendo a 4 lobectomías en el lóbulo inferior izquierdo por bronquiectasias e infecciones a repetición; además, se colocaron colleras en oído a dos pacientes.
La confirmación diagnóstica mediante biopsia de cilios se realizó en 36 pacientes, resultando que el 100% de ellos tenía alteraciones de los brazos de dineína, siendo ésta la alteración genética más frecuente. 28% de estas alteraciones además estaban asociadas a otros defectos microtubulares y dos pacientes presentaban síndrome de Kartagener clásico (situs inversus, sinusitis, bronquiectasias). No se realizó microscopía por falta de tamańo muestral; además, uno de los pacientes tenía el antecedente de un hermano con disquinesia ciliar primaria confirmada.

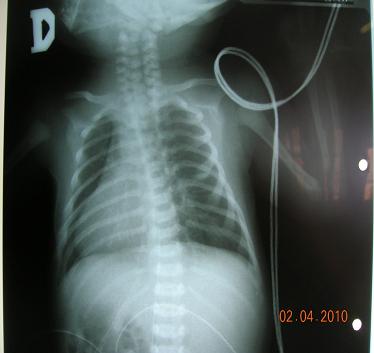 Figura 1. Radiografía de tórax que muestra dextrocardia, e infiltrado intersticial bilateral, en RN a los 6 días de vida.
Figura 1. Radiografía de tórax que muestra dextrocardia, e infiltrado intersticial bilateral, en RN a los 6 días de vida.
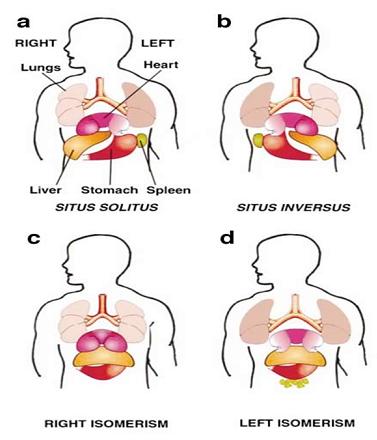 Figura 2. Se muestran esquemáticamente las alteraciones morfológicas correspondientes a las diversas alteraciones de la lateralidad, como situs solitus, situs inversus, isomerismo derecho e izquierdo.
Figura 2. Se muestran esquemáticamente las alteraciones morfológicas correspondientes a las diversas alteraciones de la lateralidad, como situs solitus, situs inversus, isomerismo derecho e izquierdo.
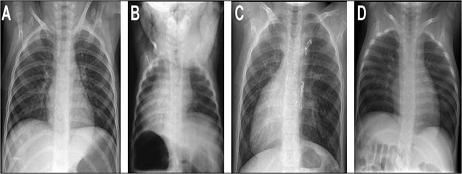 Figura 3. La imagen “A” corresponde a una radiografía de tórax normal, la imagen “B” a un situs inversus total, la imagen “C” a un isomerismo derecho y la imagen “D” un isomerismo izquierdo.
Figura 3. La imagen “A” corresponde a una radiografía de tórax normal, la imagen “B” a un situs inversus total, la imagen “C” a un isomerismo derecho y la imagen “D” un isomerismo izquierdo.
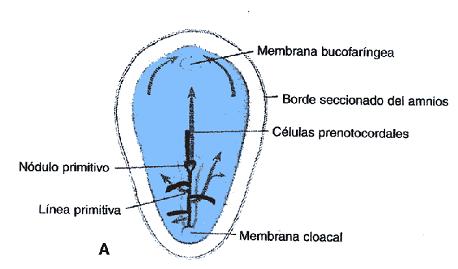 Figura 4. Esquema embrionario donde se muestra el nodo de Hensen y línea primitiva.
Figura 4. Esquema embrionario donde se muestra el nodo de Hensen y línea primitiva.
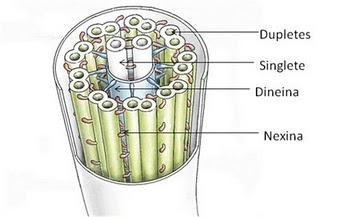 Figura 5. Esquema que ilustra modelo de cilio conformado por microtúbulos “9+2”.
Figura 5. Esquema que ilustra modelo de cilio conformado por microtúbulos “9+2”.
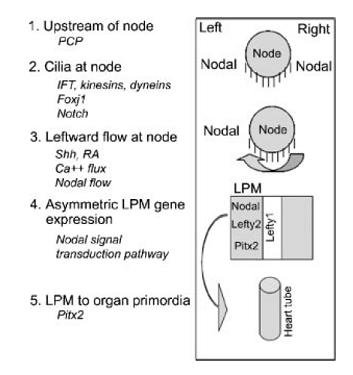 Figura 6. Seńales en el nodo que participan en la estructura y función ciliar.
Figura 6. Seńales en el nodo que participan en la estructura y función ciliar.
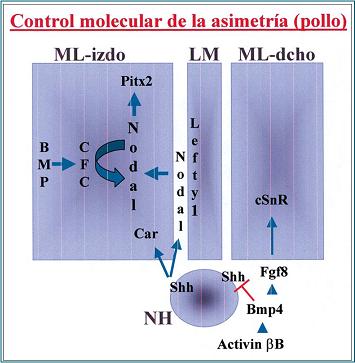 Figura 7. Cascadas de mediadores relacionados a la asimetría.
Figura 7. Cascadas de mediadores relacionados a la asimetría.
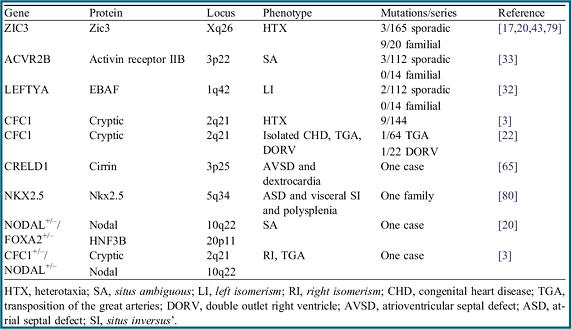 Figura 8. Genes relacionados a las asimetrías (11).
Figura 8. Genes relacionados a las asimetrías (11).
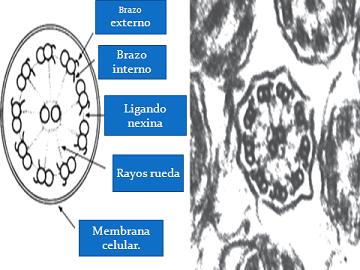 Figura 9. Microscopía electrónica de cilio “9+2” (15).
Figura 9. Microscopía electrónica de cilio “9+2” (15).
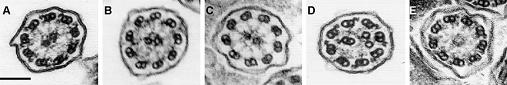 Figura 10. “A” cilio normal, “B” ausencia de brazos de dineína externos, “C” ausencia total de brazos de dineína (internos e internos), “D” ausencia de brazos de dineína internos y desorganización, “E” ausencia de complejo central (17).
Figura 10. “A” cilio normal, “B” ausencia de brazos de dineína externos, “C” ausencia total de brazos de dineína (internos e internos), “D” ausencia de brazos de dineína internos y desorganización, “E” ausencia de complejo central (17).
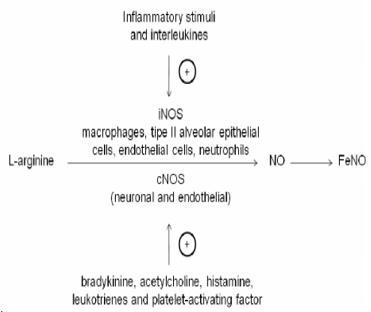 Figura 11. Mediadores de la producción de óxido nítrico en el epitelio respiratorio, a partir de L-arginina.
Figura 11. Mediadores de la producción de óxido nítrico en el epitelio respiratorio, a partir de L-arginina.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el marco de las reuniones clínicas del Servicio de Pediatría del Complejo de Salud San Borja-Arriarán. La publicación de estas actas científicas ha sido posible gracias a una colaboración editorial entre Medwave y el Servicio de Pediatría. El jefe de Servicio es el Dr. Francisco Barrera y el coordinador de las Reuniones Clínicas es el Dr. Luis Delpiano.
Expositora:
Carolina Sugg Herrera[1]
Citación: Sugg C. Alterations of left-right asymmetry and primary ciliary dyskinesia. Medwave. 2010;10(9):e4772 doi: 10.5867/medwave.2010.09.4772
Fecha de publicación: 1/10/2010
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión







 Estudios originales
Estudios originales