Resumen
La publicaci¾n de estas Actas CientĒficas ha sido posible gracias a una colaboraci¾n editorial entre Medwave y el Servicio de PediatrĒa del Hospital ClĒnico San Borja Arriarßn. Edici¾n cientĒfica: Dr. Luis Delpiano.
Historia
En el siglo II antes de Cristo, el patriarca judĒo Rabbi Judah estableci¾ una norma que eximĒa de la circuncisi¾n al tercer hijo de una mujer, en caso de que dos de sus hermanos mayores hubieran muerto o sufrido grandes hemorragias despuķs de este procedimiento. Sin embargo, las primeras descripciones cientĒficas de la hemofilia datan de fines del siglo XVIII, momento en el que algunos autores describieron familias en las cuales los varones sufrĒan hemorragias postraumßticas anormalmente prolongadas. Luego, en el siglo XIX se acumul¾ una extensa literatura sobre esta enfermedad hemorrßgica, a la que se adjudicaron distintos nombres, tales como hemorrea, idiosincrasia hemorrßgica, hematofilia y dißtesis hemorrßgica hereditaria, hasta que finalmente adquiri¾ su nombre actual: hemofilia, que significa amor a la sangre, tķrmino que dio el tĒtulo a un extenso tratado publicado en 1828. El compromiso de las articulaciones, que es el sĒntoma mßs caracterĒstico de la hemofilia, se describi¾ con detalle reciķn en 1890; antes se confundĒa con artritis reumßtica, tuberculosa o de otros tipos. En la realeza europea hubo varios casos de hemofilia, de los cuales el mßs conocido es el de Alexei Romanov, hijo del ·ltimo zar de Rusia, Nicolßs II (Fig. 1).
Figura 1. El Zarevich Alexei Romanov era hemofĒlico.
Hacia 1938 los investigadores descubrieron que s¾lo la transfusi¾n de sangre era una terapia eficaz; en los a±os 50 se desarrollaron las tķcnicas para preparar factor VIII humano en Gran Breta±a, Francia y Suecia; tambiķn en esa ķpoca se desarrollaron concentrados a partir de sangre de animales. En 1965 se adopt¾ en forma amplia el uso de crioprecipitado en los servicios de terapia transfunsional, como tratamiento sustituto del factor antihemofĒlico para los procedimientos de rutina. Desde entonces, los concentrados liofilizados han mejorado la vida de los hemofĒlicos de tal manera, que hoy los pacientes se pueden autotratar en forma precoz mediante inyecci¾n
endovenosa de los preparados de factor VIII/IX, que pueden conservar en sus hogares, lo que ha permitido tratar y prevenir las hemorragias. En la actualidad existen ademßs los preparados recombinantes, que evitan el riesgo de transmisi¾n de enfermedades, pero los liofilizados extraĒdos del plasma humano tienen una excelente bioseguridad.
Definici¾n, cuadro clĒnico, patogenia
En Chile, a partir del primero de julio del 2006 la hemofilia se incorpor¾ al plan AUGE ¢ GES
(Acceso Universal al Sistema de GarantĒas ExplĒcitas en Salud), para lo cual se utiliz¾ la siguiente definici¾n: ōEnfermedad hereditaria, ligada al sexo, congķnita, caracterizada por sangrado excesivo con tendencia a lo incoercible, sin que exista relaci¾n causa efecto evidente, que se produce en cualquier sitio de la economĒa, a menos que el paciente reciba en forma oportuna, en cantidad y calidad, el o los factores deficientes por vĒa intravenosa, a lo largo de toda su vida, ya sea en forma de demanda precoz o preventiva. Las secuelas y complicaciones biopsicosociales de esta enfermedad, sin manejo oportuno con calidad, cantidad y seguridad biol¾gica, son causa de gran carga de enfermedad sĒquica, fĒsica o ambas, de carßcter invalidante y muerte prematuraö.
Las hemofilias, que se clasifican como enfermedades hereditarias de la coagulaci¾n, caracterizadas por una alteraci¾n en la hemostasia secundaria, pueden ser de tres tipos:
- Hemofilia A, que es la mßs frecuente y se debe a dķficit de factor VIII;
- Hemofilia B, que se debe a dķficit de factor IX;
- Hemofilia C, que se caracteriza por dķficit de factor XI (muy rara; en Chile se han comunicado s¾lo dos casos).
Los sĒntomas de la hemofilia estßn dados por las hemorragias que se producen en los diferentes sistemas:
- Hemartrosis: es el sĒntoma mßs caracterĒstico de la hemofilia. Se produce cuando los ni±os empiezan a caminar, alrededor del a±o de edad.
- Hematomas profundos.
- Hemorragia quir·rgica.
- Alveolorragia.
- Epistaxis.
- Equimosis.
- Hematuria: se presenta por lo menos una vez al a±o.
- Hemorragia intracraneana: es la principal causa de muerte en los pacientes hemofĒlicos. El traumatismo desencadenante puede ser de bajo impacto.
- Hemorragia del frenillo.
- Hemorragia digestiva.
- Hemorragia en sitios de vacuna, que puede ser muy precoz, por ejemplo, al administrar la vacuna BCG.
La hemartrosis clßsica de rodilla se caracteriza por pķrdida de las lĒneas articulares y de su funci¾n (Fig. 2). La ·nica alternativa es el uso de pr¾tesis en la edad adulta, porque la hemorragia produce gran da±o articular, que conduce a la invalidez en pacientes que recibieron un tratamiento inadecuado durante su infancia (Fig. 3). Lo mismo ocurre en otras articulaciones, como cadera, que tambiķn podrĒa requerir pr¾tesis. Estas cirugĒas tienen un gran impacto econ¾mico, ya que se requiere una cantidad considerable de factor VIII durante la cirugĒa y una rehabilitaci¾n adecuada, para que el paciente no sangre. Cualquier articulaci¾n podrĒa sangrar, a excepci¾n de la temporomandibular, que raras veces presenta este problema.
Figura 2. Hemartrosis de rodilla derecha.
Figura 3. Pr¾tesis en ambas rodillas por secuelas de hemartrosis.
Con respecto a su frecuencia, la hemofilia A se presenta en 1 de cada 10.000 nacidos vivos y la hemofilia B, en 1 de cada 30.000; en 30% de los casos no existen antecedentes familiares y se denominan primer mutante o hemofĒlicos de novo.
En cuanto a la patogenia, los genes de los factores VIII y IX se localizan en el cromosoma X, por lo tanto es una enfermedad con herencia ligada al sexo. 50% de los casos de hemofilia A grave resultan de la inversi¾n de una secci¾n del brazo largo del cromosoma X. En los pacientes con hemofilia sin antecedentes familiares pueden existir mutaciones puntuales o deleciones parciales o totales del gen, que afectan la regulaci¾n de ķste.
Clasificaci¾n
Las hemofilias se clasifican en leves, moderadas y graves seg·n el porcentaje del factor correspondiente (Tabla I). Es leve si hay entre 5% a 25% del factor; moderada si hay entre 1% a 4% y grave, si hay menos de 1% del factor. Esta clasificaci¾n se aplica a las hemofilias A y B. Las hemofilias graves sangran en forma espontßnea, incluso se producen hemartrosis espontßneas; las moderadas sangran frente a accidentes o contusiones y las leves sangran frente a procedimientos quir·rgicos.
Tabla I. Clasificaci¾n de gravedad de la hemofilia.
En el a±o 2006 se realiz¾ una estratificaci¾n etaria de las personas con hemofilia en Chile y se encontr¾ que la mayorĒa eran j¾venes: 77,4% eran menores de 40 a±os y 45,5%, menores de 20, lo que se debe a que estos pacientes mueren en edades tempranas o llegan a la adultez en muy malas condiciones, situaci¾n que se espera revertir con la incorporaci¾n de esta patologĒa al AUGE.
Manejo de las hemofilias
El manejo se puede dividir en profilaxis y manejo del sangrado agudo. El hemofĒlico que consulta en urgencia no puede esperar, debe ingresar en forma inmediata porque se puede complicar rßpidamente. El tratamiento consiste en terapia de reemplazo, que se puede efectuar mediante tres tipos de elementos:
- Liofilizados: son el tratamiento de elecci¾n. Estßn disponibles desde hace 30 a±os, pero llegaron a Chile en forma masiva hace s¾lo una dķcada. Se pueden obtener de donantes o recombinantes; estos ·ltimos son muy caros, por eso en Chile se usa el proveniente de donante. Entre sus ventajas estß que su volumen es peque±o; son de fßcil y rßpida administraci¾n, incluso se pueden autoadministrar; y son fßciles de guardar y transportar. Son de uso exclusivo endovenoso y se deben administrar alrededor de 100 unidades/minuto, no mßs rßpido.
- Crioprecipitados: son la segunda elecci¾n para los pacientes con hemofilia A. Cada bolsa contiene entre 80 y 100 unidades de factor VIII y de factor Von Willebrand.
- Plasma: se usa en caso de no existir liofilizado o crioprecipitado. Aporta 1 unidad/ml de factor.
El cßlculo de la terapia de reemplazo es sencillo: se calcula el peso del paciente y el nivel del factor que se desea obtener, porque no es lo mismo una hemorragia cerebral que una hemartrosis o una gingivorragia. Frente a una hemorragia cerebral se tendrĒa que llevar el nivel del factor a 80-100%, en cambio en una gingivorragia bastarĒa con llevarlo a 40%. En la figura 4 se muestran las f¾rmulas que se utilizan para hacer el cßlculo de la cantidad de liofilizado que se debe administrar en las hemofilias A y B. Es importante no eliminar los sobrantes del liofilizado, porque es preferible proporcionar al enfermo una cantidad mayor de concentrado.
Figura 4. Cßlculo de la terapia de reemplazo en las hemofilias A y B.
Las Normas de manejo clĒnico de las hemofilias fueron confeccionadas por un panel de expertos; actualmente estßn en revisi¾n y la nueva versi¾n deberĒa salir a mßs tardar a principios de 2007. Hasta el momento se han distribuido a los equipos de salud de la mayorĒa de los hospitales y a los enfermos, 1.500 ejemplares en formato bolsillo y 1.500 afiches con el algoritmo de manejo rßpido, en el que se destaca que la hemofilia es una emergencia permanente. Ademßs existe una credencial de identificaci¾n oficial de la condici¾n de portador de hemofilia, que deberĒa ser usada por todos los pacientes que padecen de esta enfermedad (Fig. 5).
Figura 5. Credencial de identificaci¾n oficial de la condici¾n de portador de hemofilia.
Profilaxis
La profilaxis se define como el aporte del factor de coagulaci¾n deficitario por vĒa endovenosa en forma anticipada, con el fin de prevenir las hemorragias. Se hace fundamentalmente a nivel pedißtrico. Como en la actualidad el liofilizado estß disponible para todos los pacientes, el objetivo es que todo ni±o hemofĒlico grave reciba liofilizado para prevenir el sangrado y sus complicaciones, como en la hemartrosis, lo que se logra con el solo hecho de mantener el nivel de factor de coagulaci¾n en mßs de 1%; para esto se debe efectuar la administraci¾n bisemanal de este preparado.
El principal fundamento de la profilaxis es que el da±o articular se puede prevenir mediante la administraci¾n continua de los factores en dķficit, para evitar que la concentraci¾n de los factores VIII y IX disminuya bajo 1%. En 1960 la Dra. Nilsson inici¾ el programa de profilaxis y en el a±o 1992 se public¾ el primer resultado, que demostr¾ que los ni±os que recibieron profilaxis tuvieron menor porcentaje de complicaciones. El segundo fundamento para el uso de profilaxis es que el tratamiento a demanda es insuficiente, porque cuando un paciente sangra un poco y se le administra una peque±a cantidad de factor, 54% de los pacientes presenta morbilidad musculoesquelķtica significativa y s¾lo alrededor de 4% tiene radiologĒa normal.
Tratamiento
No se debe esperar a que aparezcan evidencias directas o indirectas del efecto de una hemorragia para tomar decisiones, porque cuando eso ocurre por lo general ya es muy tarde para prevenir las complicaciones y secuelas. Si el paciente refiere un traumatismo, se debe tratar; el manejo dependerß del sitio de sangrado, ya que con este antecedente se calculan los dĒas y la cantidad de liofilizado a administrar. Ademßs de liofilizado, crioprecipitado y plasma, existe el tratamiento con coadyuvantes: desmopresina, antifibrinolĒticos (ßcido tranexßmico) y corticoides.
La desmopresina es un anßlogo sintķtico de la vasopresina, que aumenta los niveles plasmßticos del factor VIII y del factor Von Willebrand en 2 a 5 veces, a partir de los dep¾sitos que existen en las cķlulas endoteliales; por lo tanto no tendrĒa utilidad en los hemofĒlicos graves, porque ķstos no tienen mßs factor. Es muy ·til en los hemofĒlicos tipo A leve y tiene menor utilidad en los pacientes con hemofilia moderada, en los que permite evitar el uso de liofilizado en sangrados menores. Lo ideal es medir la cantidad del factor antes y despuķs de la administraci¾n de desmopresina; el nivel deseado se alcanza a los 15 minutos de administraci¾n, con una dosis de 0,3 a 0,4 ug/kg/dĒa. La dosis se debe diluir en 50 a 100 ml de suero fisiol¾gico y se debe infundir en 20 a 30 minutos, para evitar reacciones adversas. Su efecto dura entre 8 y 10 horas. La desmopresina de aplicaci¾n nasal no tiene efecto, porque la dosis es insuficiente: se requerirĒan 150 microgramos en un ni±o y 300 microgramos en un adulto.
Los antifibrinolĒticos son fßrmacos cuya acci¾n es bloquear la retroalimentaci¾n circular, que consiste en la conversi¾n de plasmin¾geno en plasmina, la consiguiente acci¾n de ķsta sobre la fibrina y la de ķsta sobre el plasmin¾geno, con su consecuente conversi¾n en plasmina. En Chile el mßs utilizado es el ßcido tranexßmico, que se utiliza en dosis de 30 a 50 mg/kg/dĒa, cada 6 a 8 horas, por vĒa endovenosa u oral. Tambiķn es muy ·til como coadyuvante en la terapia de los hemofĒlicos.
Complicaciones
Las complicaciones mßs graves son las que ocurren en el sistema nervioso central. Toda cefalea en un paciente con hemofilia se debe considerar como un
proceso expansivo intracraneano hemorrßgico hasta que se demuestre lo contrario, de modo que antes de realizar una tomografĒa axial computarizada o una resonancia magnķtica (RM) se debe administrar factor al paciente. 70% de las muertes en personas con hemofilia se deben a una hemorragia intracraneana. En estos casos el tratamiento es de urgencia y el manejo consiste en administrar una dosis de ataque, con el objetivo de aumentar el factor en un rango entre 80 a 100% y aplicarlo tanto si el tratamiento es mķdico como quir·rgico. Es importante recordar que no siempre existe el antecedente de trauma, sobre todo en los ni±os, pero basta la sospecha clĒnica para iniciar la terapia de reemplazo de inmediato; que los exßmenes complementarios deben ser exclusivamente no invasivos; y que los eventuales traslados a centros de mayor complejidad se efectuarßn s¾lo despuķs de que se haya iniciado la terapia de reemplazo.
Otra complicaci¾n de la hemofilia es el
hematoma del psoas, que se puede confundir con una apendicitis aguda, pero hoy la ecografĒa ha facilitado su diagn¾stico. Tambiķn estß el
pseudotumor hemofĒlico, que habitualmente se debe a una hemorragia compacta del subperiostio que destruye hueso y cartĒlago, dando el aspecto de un verdadero tumor; el diagn¾stico se realiza por medio de RM, es de muy difĒcil manejo y siempre debe ser sometido a biopsia. Tambiķn la
hematuria constituye una complicaci¾n, que se maneja con reposo, aumento de la ingesta de lĒquido y eventual uso de corticoides; mejora en dos a tres dĒas y no se deben usar liofilizados ni antifibrinolĒticos, porque puede producir una coagulaci¾n intrarrenal. Finalmente, estos pacientes tienen mayor Ēndice de
suicidio que la poblaci¾n general, ya que ven c¾mo se va deteriorando su calidad de vida y se deprimen al ver que no pueden jugar ni tener las mismas actividades que sus pares. Para evitar estas complicaciones, el manejo del paciente hemofĒlico debe ser multiprofesional, con participaci¾n de hemat¾logo, traumat¾logo, kinesi¾logo, terapeuta ocupacional, dentista al menos una vez al a±o y psic¾logo.
En los ·ltimos diez a±os ha habido un aumento importante en la provisi¾n de factores anti-hemofĒlicos. En 1995 prßcticamente no estaban disponibles en Chile, pero en el a±o 2006 la cantidad de liofilizado disponible aument¾ a mßs del doble, con un total de 2,49 UI/habitante/paĒs/a±o. En los registros de mortalidad disponibles entre 1980 y 2006 se encontr¾ 74 pacientes fallecidos; la hemorragia intracraneana y el SIDA causaron 71% de las muertes, pero el SIDA deberĒa disminuir a medida que se masifica el uso del liofilizado. Por otro lado, 62% de los pacientes fallecieron antes de los 40 a±os de edad y 24% fallecieron antes de los 20 a±os.
El desarrollo de
inhibidores, que son anticuerpos contra factores, es actualmente la complicaci¾n mßs grave derivada del uso de la terapia de reemplazo, en especial, de concentrados de factor antihemofĒlico. Los inhibidores se pueden desarrollar no s¾lo en pacientes hemofĒlicos, sino tambiķn en el transcurso de otras enfermedades, dando origen a la entidad que se conoce como ōhemofilia adquiridaö. La sospecha es principalmente clĒnica y se plantea cuando hay mala respuesta al tratamiento habitual o aumento de eventos en ni±os que se mantienen con profilaxis. El estudio actual de la hemofilia estß centrado en la investigaci¾n de estos anticuerpos.
En Chile hay s¾lo dos o tres pacientes con inhibidores, porque la mayorĒa de los hemofĒlicos ha tenido muy poco contacto con los factores VIII y IX, pero a medida que aumente el uso de liofilizado va a aumentar tambiķn esta complicaci¾n, que ocurre con los liofilizados recombinantes y con los provenientes de donantes. Los pacientes con tĒtulos bajos se pueden tratar con dosis mßs altas de liofilizado, mientras que los que tienen tĒtulos altos deben usar factor VII activado, que se salta parte de la cascada de coagulaci¾n, pero tiene un costo elevado: la ampolla cuesta alrededor de 500.000 pesos.
En el futuro se espera que estķ disponible la terapia recombinante, ya empleada en otros paĒses y que no presenta riesgo de transmisi¾n de enfermedades. Ademßs, en los pr¾ximos 50 a±os esperamos que la terapia gķnica alcance el desarrollo necesario para tratar esta enfermedad.

 Figura 1. El Zarevich Alexei Romanov era hemofĒlico.
Figura 1. El Zarevich Alexei Romanov era hemofĒlico.
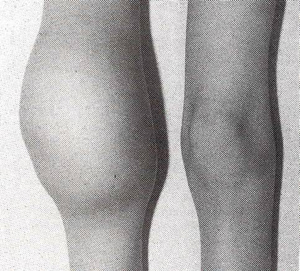 Figura 2. Hemartrosis de rodilla derecha.
Figura 2. Hemartrosis de rodilla derecha.
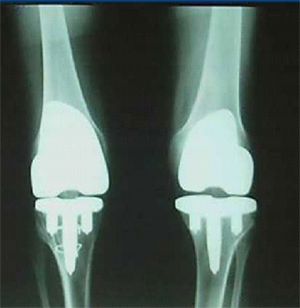 Figura 3. Pr¾tesis en ambas rodillas por secuelas de hemartrosis.
Figura 3. Pr¾tesis en ambas rodillas por secuelas de hemartrosis.
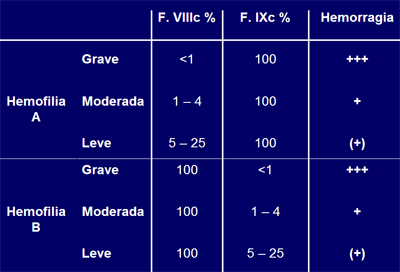 Tabla I. Clasificaci¾n de gravedad de la hemofilia.
Tabla I. Clasificaci¾n de gravedad de la hemofilia.
 Figura 4. Cßlculo de la terapia de reemplazo en las hemofilias A y B.
Figura 4. Cßlculo de la terapia de reemplazo en las hemofilias A y B.
 Figura 5. Credencial de identificaci¾n oficial de la condici¾n de portador de hemofilia.
Figura 5. Credencial de identificaci¾n oficial de la condici¾n de portador de hemofilia.

Esta
obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.








 Estudios originales
Estudios originales