Revista Biomķdica Revisada Por Pares

 Para Descargar PDF debe Abrir sesi¾n.
Para Descargar PDF debe Abrir sesi¾n.
Este texto completo es la transcripci¾n editada y revisada de una conferencia dictada en el marco de las reuniones clĒnicas del Servicio de PediatrĒa del Complejo de Salud San Borja-Arriarßn. La publicaci¾n de estas actas cientĒficas ha sido posible gracias a una colaboraci¾n editorial entre Medwave y el Servicio de PediatrĒa. El jefe de Servicio es el Dr. Francisco Barrera y el coordinador de las Reuniones ClĒnicas es el Dr. Luis Delpiano.
Las vasculitis constituyen un grupo heterogķneo e infrecuente de enfermedades que se caracterizan por presentar infiltraci¾n celular inflamatoria, que puede estar constituido por leucocitos polimorfonucleares, eosin¾filos y/o cķlulas mononucleares; y necrosis a nivel de la pared vascular. La gravedad de las vasculitis se relaciona con el tama±o, sitio y el n·mero de vasos afectados.
El da±o de la pared vascular se puede clasificar seg·n comprometa vasos grandes, medianos o peque±os; cuya sintomatologĒa es variables y se caracteriza por compromiso neurol¾gico en forma de parestesias y dolor, trombosis, hemorragias, formaci¾n de aneurismas y necrosis.
En el a±o 2005 se public¾ un nuevo criterio por la Sociedad Europea de ReumatologĒa y que defini¾ a las vasculitis seg·n el tama±o del vaso afectado y su origen primario o secundario a otras enfermedades (1). La Tabla 1 ordena las vasculitis en primarias y secundarias junto a sus entidades mßs representativas.
Tabla 1. Clasificaci¾n de las vasculitis.
Las vasculitis son poco frecuentes en ni±os, con una incidencia anual bajo los 17 a±os de edad de 20,4/100.000. Dentro de las vasculitis primarias mßs prevalentes en pediatrĒa destacan el p·rpura de Sch÷nlein Henoch (PSH), la enfermedad de Kawasaki (EK) y la arteritis de Takayasu (AT).
Factores humorales. Para explicar la etiopatogenia de las vasculitis se plantean varias hip¾tesis, como la participaci¾n de factores humorales entre los cuales se han demostrado anticuerpos especĒficos ANCA asociados a vasculitis. Estos anticuerpos activan neutr¾filos, los que determinan la inflamaci¾n; sin embargo, la falta de relaci¾n entre actividad de la enfermedad y los tĒtulos de anticuerpos sugieren la existencia de factores adicionales en la mediaci¾n del da±o vascular. Tambiķn se han detectado anticuerpos anticķlulas endoteliales, pero se desconocen si son marcadores de la enfermedad
Complejos inmunes. La otra etiopatogenia causal de vasculitis son los complejos inmunes. El tama±o e inmunorreactividad de los complejos inmunes ayudarĒan a explicar aspectos de la patogenia del PSH y de la vasculitis por crioglobulinas. Tambiķn explicarĒan la patogenia de poliarteritis nodosa (PAN) asociada al los virus de hepatitis B y C
Factores celulares. Tambiķn participarĒan linfocitos T que son atraĒdos al endotelio da±ado o infectado, lo que contribuye a aumentar la inflamaci¾n de la pared vascular mediante citotoxicidad directa o liberaci¾n de citoquinas inflamatorias. A este grupo corresponderĒan las vasculitis de tipo granulomatoso como la AT y la enfermedad de Wegener. La predilecci¾n de las distintas vasculitis sobre distintos ¾rganos de la anatomĒa se desconoce, sin embargo, pareciera que dependerĒa del antĒgeno que desencadena el fen¾meno y de variaciones regionales de los receptores de superficie.
Vasculitis leucocitoclßstica de vasos peque±os producida por complejos inmunes de tipo inmunoglobulinas A (IgA) que act·an en la activaci¾n de la vĒa alterna del complemento. Se caracteriza por un p·rpura palpable no trombocitopķnico (de preferencia en extremidades inferiores), dolor abdominal tipo c¾lico, eventuales hemorragias gastrointestinales, artritis y compromiso renal, que es la mayor causa de morbilidad, y puede progresar a insuficiencia renal cr¾nica en s¾lo el 1-3% de los casos. En raras ocasiones se producen lesiones del sistema nervioso central (SNC) o de ¾rganos respiratorios lo que se manifiesta por hemorragias . No es necesario realizar la biopsia para el diagn¾stico, ya que el cuadro clĒnico es bastante caracterĒstico.
La IgA tiene un rol importante en el PSH, cuya hip¾tesis se basa en la presencia de dep¾sitos de IgA en las lesiones vasculares; aunque ademßs se ha detectado aumento de IgA sķrica durante la fase aguda de la enfermedad y una proporci¾n de pacientes presentan complejos inmunes circulantes de tipo IgA y crioglobulinas. Algunos estudios han mostrado presencia de ANCA tipo IgA en PSH.
En una revisi¾n de casos de PSH, realizada en el hospital Roberto del RĒo, comparados con los resultados obtenidos por Cassidy, se observ¾ que el compromiso cutßneo purp·rico se presento en la totalidad de los casos en ambos estudios, seguido en frecuencia por las artralgias/artritis y el dolor abdominal. Otros hallazgos clĒnicos abarcaron microhematuria, hematuria macrosc¾pica y proteinuria, los que determinaron la presencia de sĒndromes nefr¾ticos o nefrĒticos. Las recurrencias en ambos grupos fue de alrededor de 30% (Tabla 2).
Tabla 2. Manifestaciones clĒnicas en PSH seg·n equipo de estudio.
Entre los factores desencadenantes de PSH se encuentran las infecciones por algunos microorganismos como Streptococcus tipo A (importante en ocasiones solicitar anti-estreptolisina), Bartonella henselae, Haemophilus parainfluenza, Parvovirus B19 y Virus Cocksakie. Ademßs se ha asociado PSH con el uso de ciertas drogas y vacunas.
El PSH es una afecci¾n que se desarrolla en brotes (entre oto±o e invierno) en ni±os peque±os, en donde el compromiso renal varĒa entre 15 y 62%. Los factores de riesgo que se asocian con frecuencia al da±o renal son la edad mayor a 4 a±os, p·rpura mantenida por mßs de un mes, disminuci¾n de los niveles del factor 13 mayor a 80% y manifestaciones gastrointestinales graves.
La mayorĒa de los ni±os con PSH se recuperan en su totalidad, ya que es una enfermedad bastante benigna, aunque la nefritis que provoca causa da±o renal cr¾nico en el 1,6 a 3,0% de los ni±os afectados.
El tratamiento del PSH consiste en reposo y manejo de dolores articulares. Cuando hay da±o renal se requiere otro tipo de tratamiento. Los esteroides no desempe±an un rol importante en el tratamiento, a excepci¾n del manejo del dolor abdominal, lo cual es la ·nica indicaci¾n para su uso, ademßs que no previenen el desarrollo de nefritis, aunque son ·tiles en su tratamiento.
Edema hemorrßgico agudo. Variante del PSH que se presenta en lactantes entre cuatro meses y dos a±os de vida. Se caracteriza por un comienzo agudo con fiebre y lesiones purp·ricas mßs edema perifķrico que compromete la cara, l¾bulos de las orejas y las extremidades, similares a hematomas. El compromiso gastrointestinal y renal es leve o inexistente. El curso de la enfermedad es benigno con remisi¾n espontßnea entre una y tres semanas, aunque pueden existir recurrencias. En la biopsia cutßnea se observa vasculitis leucocitoclßstica en 10 a 35% de los casos.
SĒndrome vasculĒtico autolimitado que afecta arterias medianas y peque±as, y que determina la formaci¾n de aneurismas coronarios en el 25% de los pacientes no tratados. Es la causa mßs frecuente de enfermedades cardĒacas pedißtricas adquiridas y puede determinar infartos miocßrdicos y estenosis tardĒa de las coronarias.
El 85% de los ni±os con EK son menores de 5 a±os. Los pacientes menores de tres meses y mayores de cinco a±os tienen mayor riesgo de formaci¾n de aneurismas coronarios. Las lesiones de estos pacientes se encuentran en distintos estados de evoluci¾n y afectan el endotelio de las arterias musculoelßsticas.
Existen criterios diagn¾sticos para la EK que indican el iniciar la sospecha en presencia de un cuadro febril inexplicable mayor de cinco dĒas en un lactante, que se acompa±e de cuatro de los siguientes cinco criterios: inyecci¾n conjuntival bilateral en 80 a 90%; cambios en mucosa orofarĒngea (labios rojos y/o fisurados, lengua color frutilla y faringe roja) en el 80 a 90%; cambios en extremidades como eritema o edema de manos o pies y descamaci¾n periungueal en 80%; rash polimorfo primariamente en tronco mayor a 90%, no vesicular; y linfadenopatĒa cervical mayor de 1,5 cm de dißmetro mayor en el 50% de los casos. Se debe sospechar siempre cuando exista fiebre inexplicable aunque no se cumplan los criterios (2) .
Los exßmenes de laboratorio son inespecĒficos. Destaca la elevaci¾n de parßmetros de inflamaci¾n sistķmica como aumento de velocidad de hemosedimentaci¾n (VHS), de la proteĒna C reactiva (PCR), de alfa 1 antitripsina, leucocitosis con desviaci¾n a izquierda y trombocitosis cercana a 1.000.000/mm3. Tambiķn puede haber anemia normocĒtica normocr¾mica, piuria de origen uretral, aumento de transaminasas y bilirrubinemia, ademßs de aumento de mononucleares en lĒquido cefalorraquĒdeo sin modificaci¾n de glucosa y proteĒnas.
El tratamiento consiste en el uso de gamma globulina endovenosa en dosis de 2g/kg en forma precoz, suministrada en forma ideal por infusi¾n en el transcurso de dos a cinco dĒas, lo que disminuirĒa la incidencia de formaci¾n de aneurismas. Tambiķn se indica Aspirina en dosis altas (30 a 50mg/Kg/dĒa), en cuatro dosis, y bolos de metilprednisolona cuando surge resistencia a la gamma globulina. En la actualidad se ha probado terapia Anti-TNF como el Infliximab que ha arrojado buenos resultados en cuadros resistentes a bolos de metilprednisolona.
Vasculitis que afecta a grandes vasos, infrecuente en Chile, pero frecuente en la poblaci¾n asißtica; con predominio en mujeres en la tercera dķcada de la vida y s¾lo 7 % de los pacientes son menores de 10 a±os. Es la tercera vasculitis por frecuencia en ni±os.
Los sĒntomas mßs comunes de presentaci¾n son cefalea (84%), dolor abdominal (37%), claudicaci¾n de extremidades (32%), fiebre (26%) y pķrdida de peso (10%). En el examen fĒsico se pesquisa hipertensi¾n arterial (89%), ausencia de pulsos perifķricos o diferencias en la amplitud de pulsos entre extremidades superiores e inferiores (58%) y soplos arteriales en cuello o abdomen (42%); raz¾n por lo cual se debe medir la presi¾n de las extremidades y buscar diferencias entre ellas (Tabla 3).
Su etiologĒa se desconoce, aunque se sugiere, por histopatologĒa e inmunoquĒmica de autopsia, que el mecanismo de da±o es por linfocitos T. Las lesiones son de tipo granulomatoso y comprometen desde la adventicia hasta la capa media. El diagn¾stico se realiza por ubicaci¾n de la arteritis que compromete la aorta y sus ramas.
El estßndar de oro en el diagn¾stico es la angiografĒa. Los criterios diagn¾sticos se basan en demostrar alteraciones angiogrßficas, ya sea por mķtodos convencionales, tomografĒa axial computada (TAC) o resonancia nuclear magnķtica de la aorta o sus ramas mayores, mßs al menos uno de los siguientes criterios: disminuci¾n de los pulsos perifķricos o claudicaci¾n de extremidades, diferencia de 10 mm de mercurio en cifras de presi¾n arterial entre las extremidades derecha e izquierdas; soplos en la aorta o sus ramas e hipertensi¾n seg·n las tablas infantiles (3).
Los mķtodos de imßgenes que se pueden aplicar para el diagn¾stico de AT son la angiografĒa (que es muy riesgosa); la angiorresonancia que resulta ·til para observar las dilataciones y estenosis de la aorta y sus ramas; y la angio TAC. Por medio de la angiografĒa se demostr¾ que las lesiones mßs frecuentes eran estenosis, oclusi¾n, dilataci¾n y aneurismas; las cuales comprometĒan con frecuencia a arterias renales, arterias car¾tidas, subclavia y aorta .
La AT es una enfermedad muy grave y de difĒcil tratamiento, aunque para el manejo se utilizan los corticoides y se puede cuantificar la evoluci¾n de los pacientes con la angiorresonancia al medir el tama±o de las lesiones. Tambiķn, al haber resistencia a corticoides, se puede utilizar anti-TNF y algunos inmunosupresores como la azatriopina, metotrexato y ciclofosfamida.
Vasculitis que produce cambios inflamatorios de tipo necrotizante lo que determina la formaci¾n de aneurismas que afectan arterias medianas y peque±as; tanto a nivel cutßneo como sistķmico. Se asocia a infecciones por hepatitis B y C, por lo que siempre se deben estudiar buscar en caso de una poliarteritis nodosa (PAN). La incidencia es mayor a los 9 o10 a±os y afecta mßs a varones. Tambiķn se asocia a la fiebre mediterrßnea familiar, por lo que habrĒa un componente genķtico asociado.
La PAN cutßnea se limita a la piel y al sistema musculoesquelķtico, y se produce despuķs de una infecci¾n farĒngea por estreptococo beta hemolĒtico. Se presenta con sĒntomas constitucionales leves acompa±ados al examen fĒsico por lĒvedo reticularis, rash maculopapular, n¾dulos cutßneos dolorosos, paniculitis, edema color cafķ y artritis de rodillas y tobillos. Al laboratorio destaca un aumento de VHS y PCR .
La PAN sistķmica se caracteriza por comprometer cualquier arteria muscular por lo cual, ademßs de los sĒntomas constitucionales, puede presentar disfunci¾n de cualquier ¾rgano. Se manifiesta con purpura palpable, lĒvedo reticularis, lesiones dķrmicas necr¾ticas, dolor abdominal, artritis, artralgias, miositis, mialgias, hipertensi¾n renovascular, dķficits neurol¾gicos como neuropatĒa perifķrica, enfermedad pulmonar y arteritis coronaria. Las manifestaciones dependerßn de la ubicaci¾n de la vasculitis.
Al laboratorio destacan anemia, leucocitosis, trombocitosis, aumento de VHS y PCR, presencia de ANCA positivo, compromiso renal con proteinuria, hematuria, aumento de creatinina y nitr¾geno ureico.
El diagn¾stico se realiza en forma principal por imagenologĒa, ya sea angiografĒa o Angiorresonancia, y la histopatologĒa cuando existe una lesi¾n cutßnea que se pueda biopsiar.
La angeĒtis primarias del SNC es una patologĒa cada vez mßs frecuentes. Los criterios diagn¾sticos propuestos por Calabrese en adultos son la presencia de dķficit neurol¾gico adquirido en forma reciente asociado a evidencias angiogrßfĒcas o histopatol¾gicas de vasculitis del SNC en ausencia de una enfermedad inflamatoria sistķmica, ya que s¾lo se pueden manifestar con sĒntomas neurol¾gicos. Estos criterios no han sido establecidos en ni±os, pero los casos reportados se ajustan a esta definici¾n.
Las caracterĒsticas clĒnicas son cefalea severa aguda (80%), dķficit neurol¾gico focal (78%), dķficit motor o hemiparesia (62%), compromiso de nervios craneanos (59%), disfunci¾n cognitiva (56%), aparici¾n de convulsiones y sĒntomas constitucionales (18% cada uno).
No existen exßmenes de laboratorio especĒficos que permitan realizar el diagn¾stico. El lĒquido cefalorraquĒdeo (LCR) puede mostrar aumento de cķlulas o proteĒnas, pero tambiķn puede ser normal, lo cual sirve para descartar infecciones. Las neuroimßgenes pueden ayudar, paro TAC es poco sensible y RNM se puede alterar en el 50 % de los casos al demostrar lesiones focales y multifocales de sustancia gris y blanca de distribuci¾n vascular. La combinaci¾n de LCR y RNM normales tiene un alto valor predictivo negativo. La angiorresonancia entrega resultados semejantes a la angiografĒa convencional, pero es menos riesgosa, y puede mostrar estenosis o tortuosidad y estenosis; no obstante presenta limitaciones para detectar lesiones de vasos distales y peque±os. La biopsia cerebral o de leptomeninges es el estßndar de oro, si bien se podrĒa realizar en forma menos agresiva, tampoco entrega mucha informaci¾n.
En un estudio de 62 ni±os con vasculitis primaria del SNC, el 32% present¾ una enfermedad progresiva y el 67,4% una no progresiva. La edad fluct·o entre 0,7 a 17,6 a±os, con un promedio entre 6 y 8 a±os. El 34% se recuper¾ en forma completa, 45% sigui¾ un curso progresivo y en el 31% se detuvo su progresi¾n. En el mismo estudio, se observ¾ que a la RNM, las lesiones se localizaron en forma mßs frecuente a nivel de la sustancia blanca (92%), de las cuales 87% fueron unilaterales y 65% unifocales. En la angiografĒa se apreci¾ que el compromiso mßs frecuente se produjo a nivel de la arteria cerebral media (62%), car¾tida interna (43%), arteria cerebral anterior (34%), y arteria cerebral posterior (24%). El compromiso vascular se caracteriz¾ por presencia de estenosis distales.
Los predictores de progresi¾n en vasculitis primarias del SNC en ni±os son la disfunci¾n neurocognitiva en el momento de presentaci¾n, presencia de lesiones parenquimatosas multifocales que comprometan sustancia gris y patrones angiogrßficos que se caracterizan por lesiones esten¾ticas vasculares bilaterales distales (4).
La biopsia cerebral en adultos es positiva s¾lo en 36% de los casos y muestra lesiones granulomatosas necrotizantes. En el ni±o tienen mejor rendimiento, y la lesi¾n corresponde a un infiltrado linfocitario que compromete peque±os vasos.
El tratamiento consiste en una terapia de inducci¾n mensual con ciclofosfamida endovenosa (500-700 mg/m▓) mßs altas dosis de prednisona por seis meses. El tratamiento de mantenci¾n se realiza con mofetil micofenolato o azatioprina durante 18 meses y dosis bajas de esteroides. Es controversial el uso de anticoagulantes y tanto el diagn¾stico como el tratamiento precoz se correlacionan con recuperaci¾n completa o casi completa.
Tambiķn existen vasculitis secundarias del sistema nervioso central debido a enfermedades del colßgeno (lupus eritematoso sistķmico, enfermedad de Sj÷gren, dermatomiositis y esclerodermia); a vasculitis sistķmicas (PAN, enfermedad de Sch÷enlin Henoch, EK y vasculitis ANCA positivas); infecciones bacterianas (Streptococcus pneumoniae, Salmonella, Mycobacterias, tuberculosis), virales (virus hepatitis C, citomegalovirus, VIH) u hongos (Cßndidas y Aspergillus); y otras enfermedades como neoplasias y enfermedad de Behcet.
Las vasculitis ANCA positiva son poco frecuentes, y abarcan la enfermedad de Wegener (EW), la poliangeitis microsc¾pica (PAM) y la enfermedad de Churg-Strauss (ECS). El ANCA se debe solicitar s¾lo en sospecha de estas enfermedades.
Enfermedad de Wegener. Poco frecuente en ni±os y se caracteriza por inflamaci¾n necrotizante granulomatosa de vasos peque±os y medianos que comprometen al sistema respiratorio alto y bajo, y al ri±¾n; lo que se denomina sĒndrome pulm¾n-ri±¾n, habitual en este tipo de vasculitis.
Las manifestaciones clĒnicas corresponden al compromiso nasal y sinusal que se presenta en el 100% de los casos, compromiso pulmonar en 87%, compromiso articular en 50%, compromiso renal y afectaci¾n ocular o cutßnea en 40% de los casos cada una. La estenosis subgl¾tica es mßs prevalente que en adultos y debe orientar la sospecha diagn¾stica de una EW al igual que la hipoacusia s·bita en un ni±o.
El diagn¾stico se la realiza por la presencia de anticuerpos ANCA PR3, los que se obtienen por inmunofluorescencia, que es mßs inespecĒfica, o por ELISA, aunque en la prßctica clĒnica se deben solicitar ambos exßmenes. La confirmaci¾n diagn¾stica se realiza por biopsia sinusal o pulmonar en las que se detectan lesiones inflamatorias vasculares granulomatosas.
PoliangeĒtis microsc¾pica. Vasculitis necrotizante de vasos peque±os, sin formaci¾n de granulomas, que compromete a ri±¾n y pulm¾n, por lo que produce glomerulonefritis focal y segmentaria, y hemorragias pulmonares. Tambiķn se caracteriza por presencia de anticuerpos ANCA positivos con actividad de mieloperoxida (MPO).
Enfermedad de Churg-Strauss. Enfermedad muy rara que se caracteriza por crisis asmßticas con compromiso pulmonar, aumento de los eosin¾filos e imßgenes pulmonares migratorias. Es una vasculitis eosinofĒlica.
El examen ANCA se justifica s¾lo en sospecha de estos tres tipos de vasculitis, aunque tambiķn tendrĒa un rol en la enfermedad inflamatoria intestinal.
 Tabla 1. Clasificaci¾n de las vasculitis.
Tabla 1. Clasificaci¾n de las vasculitis.
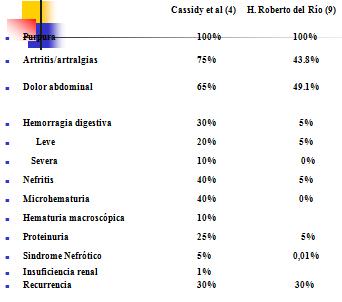 Tabla 2. Manifestaciones clĒnicas en PSH seg·n equipo de estudio.
Tabla 2. Manifestaciones clĒnicas en PSH seg·n equipo de estudio.
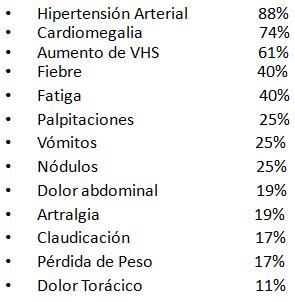 Tabla 3. SĒntomas y signos mßs frecuentes de AT en adultos. Seg·n la ubicaci¾n del aneurisma o estenosis se pueden producir el compromiso renal, si se afecta la arteria renal, o sĒntomas neurol¾gicos si compromete car¾tidas.
Tabla 3. SĒntomas y signos mßs frecuentes de AT en adultos. Seg·n la ubicaci¾n del aneurisma o estenosis se pueden producir el compromiso renal, si se afecta la arteria renal, o sĒntomas neurol¾gicos si compromete car¾tidas.
 Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripci¾n editada y revisada de una conferencia dictada en el marco de las reuniones clĒnicas del Servicio de PediatrĒa del Complejo de Salud San Borja-Arriarßn. La publicaci¾n de estas actas cientĒficas ha sido posible gracias a una colaboraci¾n editorial entre Medwave y el Servicio de PediatrĒa. El jefe de Servicio es el Dr. Francisco Barrera y el coordinador de las Reuniones ClĒnicas es el Dr. Luis Delpiano.
Expositora:
Liana Schlesinger[1]
Citaci¾n: Schlesinger L. Vasculitis of the child. Medwave 2010 Jun;10(6):e4597 doi: 10.5867/medwave.2010.06.4597
Fecha de publicaci¾n: 1/6/2010
Nos complace que usted tenga interķs en comentar uno de nuestros artĒculos. Su comentario serß publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la direcci¾n editorial considera que su comentario es: ofensivo en alg·n sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas polĒticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisi¾n por pares.
A·n no hay comentarios en este artĒculo.
Para comentar debe iniciar sesi¾n
Medwave publica las vistas HTML y descargas PDF por artĒculo, junto con otras mķtricas de redes sociales.
Dedeoglu F, Sundel RP. Vasculitis in children. Rheum Dis Clin North Am 2007;33:555-583. | CrossRef | PubMed |Spalding SJ. Kawasaki disease. MMWR Morb Mortal Wkly Rpt 1990;39:27-8. 






 Estudios originales
Estudios originales