Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Palabras clave: medicina molecular, actualización, practica clínica
Este texto completo es la transcripción editada y revisada de una conferencia dictada en reunión clínica del Departamento de Medicina, Hospital Clínico Universidad de Chile. El director del Departamento de Medicina es el Dr. Alejandro Cotera y la coordinadora de las reuniones clínicas es la Dra. Miriam Alvo.
En esta conferencia se mostrarán las aplicaciones de la genética en la práctica clínica, especialmente en las denominadas enfermedades raras.
Las enfermedades raras (ER) son las que afectan a menos de una de cada 2.000 personas (OMS); muchas afectan a una de cada 100.000 personas o aún menos que eso. Tomando en cuenta esa definición existen entre 5.000 y 8.000 ER diferentes que afectan a alrededor de 6% a 8% de la población, lo que según un cálculo de EURORDIS, la asociación de ER de Europa, corresponde a entre 24 y 36 millones de personas de la Comunidad Europea, cifra equivalente a la población de los Países Bajos, Bélgica y Luxemburgo. Debido a su baja frecuencia no se diagnostican o el diagnóstico se hace en forma tardía y muchas veces no hay un tratamiento específico o hay gran dificultad para acceder a éste.
Las ER son desconocidas incluso por la comunidad médica, por ello los pacientes afectados tienen historia de múltiples consultas a diferentes especialistas; el acceso a procedimientos diagnósticos es difícil o no existen las herramientas en el país y es frecuente que la enfermedad se asocie a discapacidad y por lo tanto, a problemas en la inserción escolar, laboral y social. Son enfermedades graves, crónicas, degenerativas, generalmente progresivas y que ponen en riesgo la vida; producen disminución de la calidad de vida y pérdida de la autonomía. Muchas veces producen dolor y sufrimiento a la persona afectada y su familia, de modo que aunque no haya tratamiento siempre se debe intentar hacer algo para mejorar la calidad y expectativa de vida.
En cuanto a su etiología, alrededor de 80% de las ER tendría un origen genético, que equivale a 3% a 4% de los nacimientos, pero habría otras causas: infecciosas, alérgicas, enfermedades degenerativas o proliferativas, que son enfermedades raras. La afección puede ser manifiesta (visible) desde el nacimiento o en la nińez, otras aparecen en la madurez, con amplia diversidad de alteraciones y síntomas que varían no sólo de una enfermedad a otra, sino también de un paciente a otro en cuanto a gravedad de las manifestaciones y evolución.
Las asociaciones de apoyo surgieron como una manera de dar soporte emocional y social; el primer grupo de apoyo fue Alcohólicos Anónimos. Actualmente estas organizaciones de apoyo son proactivas, ya no se sienten víctimas o sujetos de investigación sino que buscan a los grupos de investigadores porque saben que los avances en el conocimiento de la enfermedad aumentan la posibilidad de que se descubran nuevos tratamientos.
Un ejemplo es la fibrodisplasia osificante progresiva (FOP), una enfermedad muy rara que afecta a una de cada 2.000.000 de personas, por lo que se estima que hay alrededor de 700 personas afectadas en el mundo. La asociación internacional que apoya a estos pacientes, IFOPA, facilitó la investigación que permitió el descubrimiento del gen causante ACVR1 hace tres ańos; se trata de un gen que participa en el desarrollo morfogenético del hueso y constituye un blanco terapéutico para la enfermedad (1). IFOPA tiene miembros en 50 países y ofrece foros de debate en Internet; Moira Liljesthröm, argentina, madre de un nińo afectado de 12 ańos creó el grupo FOP Latinoamérica (ALAFOP).
Actualmente existen varias organizaciones; en Europa: EURORDIS (Organización Europea de ER) y Orphanet; en los Estados Unidos: NORD (Organización Nacional de ER) y Genetic Alliance; en Latinoamérica la Fundación Geiser y en Chile se está formando CEMINER (Centro de Manejo Integral de Enfermedades Raras) en el Hospital Clínico de la Universidad de Chile, cuyo objetivo es entregar asistencia de médicos especializados y calificados para el diagnóstico clínico de pacientes previa firma de consentimiento informado y asegurando la confidencialidad. CEMINER entrega asesoría sobre exámenes o pruebas de investigación con utilidad diagnóstica y ayuda en la interpretación de resultados de estudios citogenéticos o genéticos moleculares; también ofrece consejo genético para los pacientes y sus familiares y ayuda a constituir agrupaciones de apoyo. En la Fig. 1 se muestran los logos de algunas de estas organizaciones.
Figura 1. Enfermedades raras: organizaciones de apoyo.
El concepto de drogas huérfanas se refiere a drogas que carecen de interés comercial porque sirven sólo para un grupo limitado de pacientes; por ello desde el ańo 1983 existe en los Estados Unidos un acta sobre drogas huérfanas que contempla la entrega de incentivos a aquellos que inviertan en investigación de drogas de uso infrecuente, luego se han alineado otros países en la Unión Europea desde el ańo 2000. La legislación se ha promulgado en otros países: en Singapur, en 1991; en Japón, en 1993; en Australia, en 1998 y en la Unión Europea en el ańo 2000.
Frente a una ER, el médico general o cualquier otro especialista debe pensar y sospechar el diagnóstico, pedir las colaboraciones que se necesiten por la habitual coexistencia de múltiples afecciones, trabajar con equipos multidisciplinarios bien integrados, guiar al paciente o a sus padres en la priorización del cronograma, establecer la urgencia de algunos exámenes o evaluaciones de especialistas y luego transmitir la noción de la labor preventiva que implican algunas intervenciones que se realizan en el momento adecuado en estos pacientes y sus familias. Con el objetivo de colaborar en la difusión de estos conceptos, la Red de Aprendizaje Digital de la Facultad de Medicina de la Universidad de Chile (www.medichi.cl) entregará desde junio del ańo 2010 la cuarta versión del curso de Actualización en Genética Clínica, un curso on line que permite una interacción permanente con preguntas y que puede ser una buena aproximación a las ER.
Los exámenes genéticos se clasifican en exámenes citogenéticos, de ADN o estudio de cromosomas y metabólicos, que analizan enzimas. A continuación se analiza la utilidad de los exámenes genéticos moleculares.
Los exámenes genéticos moleculares ayudan al médico entregar la mejor atención posible a un paciente con una enfermedad hereditaria. También ayudan al individuo portador de una enfermedad hereditaria a tomar decisiones personales informadas, a través de:
Los estudios genéticos moleculares sirven también para certificar el diagnóstico en personas sintomáticas; por ejemplo, en un paciente con deterioro mental, el estudio molecular permite certificar que se trata de un corea de Huntington; y en otro caso, el examen de MLH1/MSH2 permite hacer el diagnóstico de cáncer colorrectal no poliposo hereditario en una persona sintomática que no cumple con todos los criterios de Amsterdam (Tabla I).
Finalmente, la mayor utilidad de estos exámenes es que permiten efectuar estudios predictivos en personas asintomáticas cuando hay un tratamiento disponible, lo que constituye el futuro de la Medicina: saber qué individuos tienen alto riesgo de desarrollar una enfermedad y cuáles son las medidas que se deben adoptar para prevenir sus consecuencias o eventualmente, modificar su evolución. Por ejemplo, se pueden desarrollar programas de vigilancia para prevenir la morbimortalidad en personas con alto riesgo de presentar cáncer colorrectal no poliposo hereditario, determinado mediante el examen de MLH1/MSH2 en una persona que no cumple con todos los criterios de Amsterdam.
Antes de hacer exámenes predictivos se debería conversar con el paciente y cada uno de los miembros de su familia acerca de todo lo relacionado con la enfermedad:
Asimismo, después de tener los resultados de los exámenes se debe profundizar en los siguientes aspectos:
El cáncer de colon no polipósico hereditario (HNPCC) se debe a una mutación en el gen de reparación de desfase de apareamiento o asociado a tumores con MSI (inestabilidad de microsatélites), lo que significa que el individuo tiene mecanismos de reparación de mala calidad o poco eficientes, por lo tanto tiene mayor riesgo de cáncer no sólo colorrectal, sino también de varios órganos, entre ellos endometrio, ovario, estómago, intestino delgado, vía hepatobiliar, vía urinaria, cerebro y piel. La presencia de esta mutación implica 80% de probabilidades de presentar cáncer de colon a los 61 ańos, 20 a 60% de riesgo de cáncer endometrial a los 46-62 ańos y alto riesgo de tener cáncer gástrico a los 56 ańos y cáncer de ovario a los 42,5 ańos.
Para el diagnóstico y estudio del HNPCC existen los criterios clínicos de Amsterdam, que se basan en relaciones familiares y edades de aparición, pero no son totalmente confirmatorios ya que 39% de las familias con mutaciones no cumplen con estos criterios. El estudio genético molecular para mutaciones germinales (hereditarias) en uno de los genes de reparación de desfase (MMR) MLH1, MSH2, MSH6 y PMS2 permite detectar a los individuos de riesgo; 90% de las mutaciones se encuentra en los genes MLH1 y MSH2, 7 a 10% en MSH6 y menos de 5% en PMS2. La presencia de una mutación pesa más que la historia familiar, pero no se recomienda realizar el estudio genético antes de los 18 ańos de edad en individuos de riesgo.
A continuación se describe un caso clínico de HNPCC. El consultante es un varón de 33 ańos de edad, procedente de Holanda donde reside su familia; trabaja en Chile desde hace cuatro ańos y es asintomático, pero tiene historia familiar de cáncer de colon, cáncer de endometrio y melanoma por línea materna: su abuelo falleció de cáncer de colon antes de los cincuenta ańos, su madre se trató por cáncer endometrial a los 35 ańos, por cáncer de colon a los 45 ańos y actualmente, a los 60 ańos, presenta un melanoma. El paciente está casado, tiene tres hijos y conoce la mutación de su madre gracias a un estudio efectuado en Holanda; su hermano y su hermana se realizaron el estudio, que demostró que no son portadores y él también desea saber su estado al respecto. El estudio genético descarta la mutación, por lo tanto se le indica controles colonoscópicos habituales (Fig. 2).
Figura 2. Caso clínico de HNPCC.
En el consejo genético de esta enfermedad se debe tomar en cuenta las siguientes consideraciones: es de herencia autosómica dominante; la mayoría de los afectados heredó la condición de un progenitor, pero no siempre existe el antecedente de cáncer (puede haber fallecimientos por otras causas antes de que éste se manifieste); cada hijo de un afectado por HNPCC tiene 50% de probabilidades de heredar la mutación; es posible realizar el diagnóstico prenatal si se ha identificado la mutación familiar, sin embargo es poco frecuente esta solicitud ante patologías del adulto que tienen un tratamiento disponible.
Otra condición hereditaria asociada a cáncer de colon son las relacionadas con el gen APC (Condiciones Asociadas a Pólipos), que se manifiestan en cuatro formas principales:
El síndrome de Gardner, que se asocia a cáncer hereditario, se caracteriza por: tumores de piel y quistes epidermales; osteomas mandibulares; pólipos colónicos con 100% de transformación a adenocarcinoma; lesiones multifocales pigmentadas del fondo de ojo; anomalías dentales y gen APC (Fig. 3).
En el diagnóstico y exámenes de las condiciones con poliposis asociada causadas por mutaciones en el gen APC, los hallazgos clínicos son prioritarios; se detectan mutaciones de APC en más de 90% de los probandos con típico FAP. Los estudios están indicados en el diagnóstico precoz de parientes en riesgo, para confirmar el diagnóstico de FAP o FAP atenuado en casos con hallazgos equívocos (menos de 100 pólipos adenomatosos).
A continuación se describe un caso clínico de APC. Hay antecedentes familiares de su madre fallecida de cáncer de colon a los 58 ańos y su abuelo materno fallecido a los 61 ańos, también de cáncer de colon, información que por lo general no se pregunta en una anamnesis. El paciente tiene 52 ańos. A los 48 ańos presenta hemorragia digestiva baja “gotas de sangre”, sin dolor y tiene episodios frecuentes de “gastritis” que automedica con Omeprazol. En 2008 presenta nuevo episodio de rectorragia (gotas) por lo que se realiza una colonoscopía, que muestra más de 400 pólipos en todos los segmentos del colon. Las biopsias revelan pólipos adenomatosos sin signos de malignidad; la endoscopía alta no tiene alteraciones. En la siguiente imagen se muestra la genealogía: el paciente de 52 ańos, su madre y su abuelo fallecido. Se hizo estudio de mutaciones del gen APC y se encontró una inserción de 17 bases pares que causaba un codón de término del exon 9 de APC. El paciente ha tenido tres esposas, tiene dos hijos con cada una de ellas y todos tienen 50% de probabilidades de haber heredado la mutación (Fig. 4).
Figura 4. Caso clínico de APC: genealogía.
En el diagnóstico diferencial de condiciones APC con poliposis se incluye: poliposis asociada a MYH; cáncer colorrectal hereditario no polipósico (HNPCC); síndrome de Turcot; síndrome de Peutz-Jeghers (PJS); síndromes PTEN: síndromes de Cowden, Bannayan-Riley-Ruvalcaba, Proteus y similar a Proteus; síndrome de poliposis juvenil (JPS); síndrome hereditario de poliposis mixta (HMPS); neurofibromatosis tipo 1(NF1); síndrome Cronkite-Canadá; hiperplasia nodular linfoide; poliposis linfomatosa; poliposis inflamatoria; tumores esporádicos colorrectales; poliposis hiperplástica.
Entre las numerosas condiciones que se deben considerar al hacer el diagnóstico diferencial con poliposis está el síndrome de Peutz-Jeghers, que se manifiesta por manchas pigmentarias en la piel y mucosas y se asocia a mayor riesgo de cáncer, no sólo gastrointestinal, sino también pancreático y uterino. En este caso el afectado es el gen STK11 (Fig. 5).
Figura 5. Síndrome de Peutz-Jeghers: manchas pigmentarias en piel y mucosas.
La enfermedad de Cowden se caracteriza por la presencia de pólipos en colon y estómago, macrocefalia, lesiones mucocutáneas y mayor riesgo de cáncer tiroideo (10%) y de mama (50%). Las mutaciones ocurren en el gen PTEN (Fig. 6).
Figura 6. Cáncer hereditario: enfermedad de Cowden lesiones mucocutáneas.
En las condiciones ligadas al gen APC se recomienda efectuar el estudio en personas que decidan por sí mismas determinar su condición y sus riesgos, ya que es una enfermedad maligna y precoz. Los estudios son:
El consejo genético en APC debe considerar que se trata de una enfermedad de herencia autosómica dominante, por lo tanto alrededor de 75 a 80% de los individuos con poliposis asociada a APC tienen un progenitor afectado y la descendencia de un afectado tiene 50% de riesgo de heredar el gen alterado APC. El diagnóstico prenatal es posible si previamente se ha identificado la mutación en un familiar afectado.
Hay una relación entre las malformaciones y los cánceres, porque los genes que participan en el desarrollo también participan en el desarrollo de cáncer. Lammi, un autor finlandés demostró en una familia que las mutaciones en el gen AXIN2 causan oligodoncia familiar y predisposición a cáncer colorrectal (2). Sería interesante investigar la posibilidad de utilizar el análisis de la dentadura como marcador precoz de cáncer de colon (Fig. 7).
El cáncer hereditario corresponde a menos de 5% de todos los cánceres, pero se debe sospechar que una familia tiene mayor riesgo de ocurrencia de cáncer hereditario en los siguientes casos:
Existen varias asociaciones de apoyo para pacientes y familias con HNPCC y APC, entre ellas: Colon Cancer Alliance (www.ccalliance.org); Genetics of Colorectal Cancer; Hereditary Colon Cancer Association (www.hereditarycc.org); American Cancer Society (www.cancer.org); Collaborative Group of the Americas on Inherited Colorectal Cancer (www.cgaicc.com); National Library of Medicine Genetics Home Reference Familial adenomatous polyposis; Colorectal Cancer Coalition (www.FightColorectalCancer.org); United Ostomy Association, Inc (www.uoa.org); PMP Pals (www.pmppals.org); AMC Cancer Research Center and Foundation (www.amc.org); CancerCare (www.cancercare.org).
El corea de Huntington es una enfermedad progresiva con alteraciones motoras, cognitivas y siquiátricas, que se presenta entre los 35 y 44 ańos de edad y se caracteriza por un tiempo medio de sobrevida de 15 a 18 ańos después del inicio. El diagnóstico se basa en historia familiar positiva, hallazgos clínicos característicos y detección de expansiones del trinucleótido citosina-alanina-guanina (CAG) en el gen HD. Los alelos normales tienen 26 o menos repeticiones de este trinucleótido: los alelos intermedios tienen 27 a 35 repeticiones y los portadores no tienen riesgo de desarrollar la enfermedad, pero pueden tener un hijo con ésta (alelos mutables); los alelos causantes de HD tienen 36 o más de estas repeticiones. Una penetrancia reducida, con 36 a 39 repeticiones, podría desarrollar menos síntomas; con penetrancia completa, 40 o más, existe gran certeza de manifestación completa de la enfermedad.
El manejo del corea de Huntington es complejo y se orienta a aliviar muchos de los síntomas, mediante el uso de neurolépticos, benzodiazepinas, agentes antiparkinsonianos, drogas psicótropas y antiepilépticos, como el ácido valproico. Además se requiere apoyo de enfermería y prevención de complicaciones secundarias, para lo cual se debe efectuar vigilancia permanente del paciente y evaluar regularmente la apariencia y gravedad del corea y la presencia de rigidez, problemas de la marcha, depresión, cambios conductuales y deterioro cognitivo. Se debe evitar el uso de fármacos que contengan L-dopa, así como el consumo de alcohol y tabaco. Es imprescindible el apoyo psicológico y educacional a nińos y adolescentes con un padre afectado.
Un caso clínico de corea de Huntington es el de un paciente varón de 33 ańos, que está recibiendo tratamiento por trastorno obsesivo compulsivo. Tiene un hijo de un ańo de edad. En el ańo 2006 comienza con bruxismo, dislalia y movimientos distónicos de la lengua, sin movimientos coreicos; tiene hemograma, función renal, cupruria, C3 y C4, ceruloplasmina y perfil hepático normales; y detección de virus hepáticos, anticuerpos antinucleares, anticuerpos antimitocondriales negativa. La CK y la deshidrogenasa láctica tienen un aumento leve. La biopsia hepática muestra un infiltrado linfoide de predominio portal. La ecografía abdominal revela esplenomegalia y la RNM cerebral, atrofia de la corteza de los núcleos caudados, baja seńal de los globos pálidos y aumento de la seńal en la cápsula externa, sugerentes de enfermedad de Wilson, por lo que se hizo biopsia hepática, que finalmente descartó esta enfermedad. Tiene una hermana de 32 ańos de edad, sin hijos, que tuvo depresión en la adolescencia, desde los 25 ańos de edad está en tratamiento por epilepsia mioclónica juvenil benigna y es portadora de ovario poliquístico con resistencia a la insulina e hipertricosis. Desde 2004 presenta bruxismo, dislalia, temblores de extremidades y mioclonías; tiene nivel de cobre normal y leve aumento de ceruloplasmina. La RNM cerebral es sospechosa de depósito mineral. La madre de ambos tiene 57 ańos y es portadora de hipertensión arterial, depresión y bruxismo. En la siguiente imagen se resume la genealogía de este paciente: se observa su hijo, la hermana mencionada, otros dos hermanos más jóvenes y la madre, que también tendría manifestaciones de la enfermedad. El estudio molecular demostró que el paciente tiene 17 repeticiones en un alelo y 44 repeticiones en el otro, por lo tanto hay certeza absoluta de que está evolucionando con las manifestaciones del corea de Huntington. Falta confirmar el diagnóstico en la hermana y la madre y en los dos hermanos, que no tienen síntomas, pero podrían desarrollarlos en el futuro (Fig. 8).
Figura 8. Genealogía de paciente con corea de Huntington.
En cuanto al consejo genético, el corea de Huntington es una enfermedad de herencia autosómica dominante que se suele manifestar a una edad en que el individuo ya tiene hijos, por lo tanto ya no puede tomar decisiones al respecto. En todo caso, hay que saber que el hijo de un probando tiene 50% de riesgo de heredar la mutación, por lo tanto el estudio predictivo en adultos asintomáticos con 50% de riesgo está disponible; no obstante, se debe meditar cuidadosamente esta decisión, incluyendo el consejo genético antes y después del examen, ya que no existe un tratamiento disponible. En individuos de riesgo menores de 18 ańos no se debería realizar estudios predictivos. El diagnóstico prenatal está disponible, sin embargo no son frecuentes las solicitudes de estudio prenatal.
Finalmente, si se hiciera tamizaje poblacional para identificar factores genéticos de riesgo habría que considerar que en caso de que las pruebas salieran positivas se debería adoptar medidas de prevención, por lo tanto el individuo debe preguntarse a sí mismo si estaría dispuesto a hacer cambios importantes en cuanto a dieta, estilo de vida, medio ambiente y uso de medicaciones, entre otros aspectos. Esto requiere de una atención médica personalizada.
Como afirma Hipócrates en su primer aforismo: “Corta es la vida, largo el camino, la ocasión fugaz, falaces las experiencias, el juicio difícil. No basta, además, que el médico se muestre tal en tiempo oportuno, sino que es menester que el enfermo y cuantos lo rodean coadyuven”.
 Figura 1. Enfermedades raras: organizaciones de apoyo.
Figura 1. Enfermedades raras: organizaciones de apoyo.
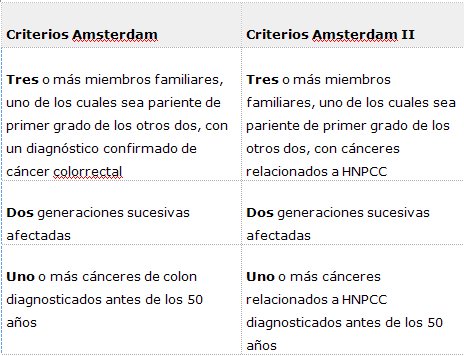 Tabla I. Criterios de Amsterdam y Amsterdam II par el diagnóstico clínico de HNPCC (cáncer de colon hereditario no poliposo).
Tabla I. Criterios de Amsterdam y Amsterdam II par el diagnóstico clínico de HNPCC (cáncer de colon hereditario no poliposo).
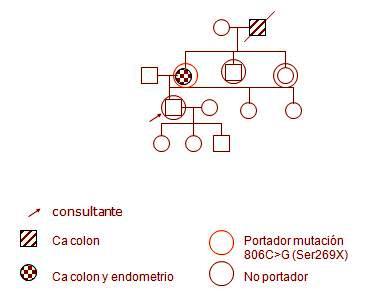 Figura 2. Caso clínico de HNPCC.
Figura 2. Caso clínico de HNPCC.
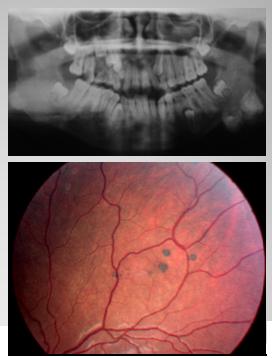 Figura 3. Lesiones multifocales pigmentadas del fondo de ojo y anomalías dentales en el Síndrome de Gardner.
Figura 3. Lesiones multifocales pigmentadas del fondo de ojo y anomalías dentales en el Síndrome de Gardner.
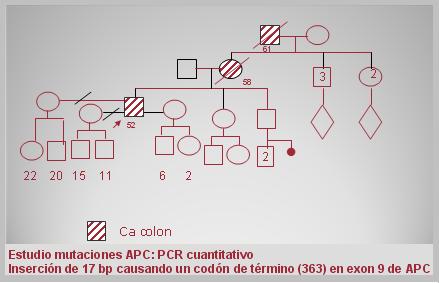 Figura 4. Caso clínico de APC: genealogía.
Figura 4. Caso clínico de APC: genealogía.
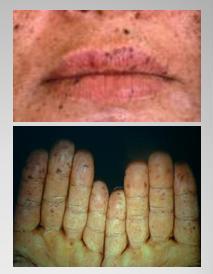 Figura 5. Síndrome de Peutz-Jeghers: manchas pigmentarias en piel y mucosas.
Figura 5. Síndrome de Peutz-Jeghers: manchas pigmentarias en piel y mucosas.
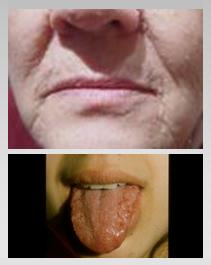 Figura 6. Cáncer hereditario: enfermedad de Cowden lesiones mucocutáneas.
Figura 6. Cáncer hereditario: enfermedad de Cowden lesiones mucocutáneas.
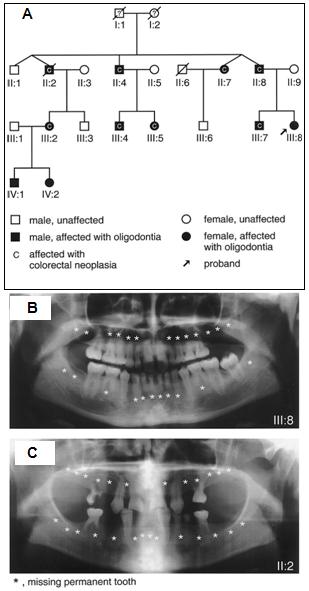 Figura 7. Mutaciones en el gen AXIN2 causan oligodoncia familiar y predisposición a cáncer colorrectal.
Figura 7. Mutaciones en el gen AXIN2 causan oligodoncia familiar y predisposición a cáncer colorrectal.
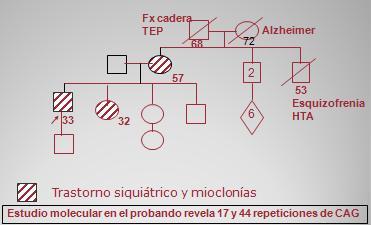 Figura 8. Genealogía de paciente con corea de Huntington.
Figura 8. Genealogía de paciente con corea de Huntington.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en reunión clínica del Departamento de Medicina, Hospital Clínico Universidad de Chile. El director del Departamento de Medicina es el Dr. Alejandro Cotera y la coordinadora de las reuniones clínicas es la Dra. Miriam Alvo.
Expositora:
Silvia Castillo Taucher[1]
Citación: Castillo S. An update in molecular medicine: applications for clinical practice. Medwave 2010 Feb;10(02):e4376 doi: 10.5867/medwave.2010.02.4376
Fecha de publicación: 1/2/2010
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006 May;38(5):525-7. Epub 2006 Apr 23.
| PubMed |






 Estudios originales
Estudios originales