Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el marco de las reuniones clínicas de la Unidad General de Cuidados del Nińo del Hospital Padre Hurtado. La publicación de estas actas científicas ha sido posible gracias a una colaboración editorial entre Medwave y la Unidad. El jefe de la UGCN es el Dr. Alejandro Donoso y el Encargado de las Reuniones Clínicas es el Dr. Mario Vildoso.
Los fármacos anticonvulsivantes se utilizan ampliamente en pediatría, aunque no son inocuos. Se ha descrito una serie de efectos colaterales asociados a este tipo de medicamentos, incluyendo efectos sobre muchos nutrientes y por tanto, sobre la condición nutricional de los pacientes que los reciben.
Los anticonvulsivantes se separan en dos grupos: antiguos y nuevos. Todos ellos actúan sobre el sistema nervioso central a distintos niveles, como canales iónicos, unión sináptica, mediadores sinápticos y conducción del impulso, entre otros, de tal manera que disminuyen las descargas eléctricas responsables de las crisis convulsivas. Sin embargo, esta acción sobre el sistema nervioso central se acompańa de una serie de efectos metabólicos diversos. Algunos de estos cambios bioquímicos se correlacionan con determinadas manifestaciones clínicas. Los antiguos anticonvulsivantes son fenobarbital, ácido valproico, carbamazepina, fenitoína y primidona. Los más usados son los tres primeros, mientras que los dos últimos se usan en las Unidades de Cuidados Intensivos (UCI) en caso de urgencia.
Las drogas anticonvulsivantes producen alteraciones importantes en el metabolismo óseo-mineral. Los anticonvulsivantes de acción prolongada tienen mayor riesgo de producir desmineralización ósea y fractura sobre hueso patológico (1), eventos que son facilitados por factores de riesgo como déficit de vitamina D, hipocalcemia e hiperparatiroidismo secundario (2). Estos efectos se describieron inicialmente para el fenobarbital, hace casi treinta ańos (3) y posteriormente, para la carbamazepina (4, 5) y son especialmente relevantes en pacientes con encefalopatía, que permanecen más tiempo postrados y, por lo tanto, tienen menor desarrollo muscular y óseo.
En la siguiente imagen se ilustra el mecanismo propuesto para el desarrollo de osteopenias mediado por el receptor Pregnano X, relacionado con la vitamina D. La figura muestra el sistema de biosíntesis de la vitamina D, especie de hormona cuyos precursores provienen de la dieta y son hidroxilados en primera instancia en el hígado, produciendo la molécula 25-Hidroxicolecalciferol (25(OH) D3) la cual es hidroxilada posteriormente en el rińón, produciendo la 1,25-dihidroxivitamina D3 (1,25(OH)2 D3), que es la forma activa a nivel intestinal, donde aumenta la absorción de calcio. Una vez que se establece la forma activa ésta se une a la proteína receptora de Vitamina D (VDR), que forma un complejo con el receptor X del ácido retinoico (RXR) lo que permite a la hormona ejercer sus funciones. Posteriormente se da comienzo al proceso de inactivación de las hormonas a través del receptor Pregnano X, que es activado en forma alterna por algunas drogas (Fig. 1).
Figura 1. Mecanismo propuesto para desarrollo de osteopenia mediada por PXR (receptor Pregnano X).
La hipótesis actual sobre el mecanismo de acción de las drogas anticonvulsivantes plantea que fenobarbital, fenitoína y carbamazepina actúan interfiriendo con la proteína VDR, que impide la transformación a la forma inactiva de la hormona; como consecuencia predomina la función del receptor Pregnano X, lo que provoca que se inactive la vitamina D y disminuya su acción sobre la absorción del calcio intestinal proveniente de la dieta. Por ello, se ha sugerido que la suplementación con vitamina D podría mejorar estas condiciones, ya que mejoraría el contenido mineral óseo en las personas portadoras de epilepsia que reciben fenitoína, primidona y fenobarbital; sin embargo, en una revisión de la base de datos Cochrane no se encontró evidencia suficiente para apoyar esta medida. Es necesario contar con estudios controlados, aleatorios y doble ciego para evaluar la utilidad de la vitamina D en la recuperación del contenido mineral óseo en pacientes usuarios de fármacos anticonvulsivantes (6).
En un estudio en adultos realizado en 1973 por Christiansen se evaluó a 226 adultos, 116 tratados y 110 controles y se encontró que el contenido mineral óseo es significativamente superior en pacientes con epilepsia que reciben suplementos de Vitamina D (7). En 1975 el mismo autor publicó un estudio efectuado en 25 nińos en el que concluyó que el suplemento de vitamina D tendría un efecto beneficioso, pero no se entregaron los valores reales de alteración del contenido mineral óseo y el grupo era muy pequeńo (8). Posteriormente no hay publicaciones que confirmen el beneficio de usar vitamina D y tampoco la dosis que se necesitaría, de modo que el uso de vitamina D es una práctica empírica.
En la siguiente imagen se muestra el fenómeno de transferencia de carbono que ocurre durante la utilización de las vitaminas B12, B6 y ácido fólico, en relación con la síntesis del ADN y la metilación del ADN. La homocisteína, que se ha relacionado con la ocurrencia de accidentes vasculares y debería ser retirada de la sangre lo más pronto posible, es transformada a metionina por la enzima metionina sintetasa, utilizando como cofactor a la vitamina B12. La carbamazepina y el ácido valproico interfieren en la conversión de homocisteína en metionina y aumentan los niveles en la sangre de homocisteína, la cual tiene efectos aterogénicos y epileptogénicos. Ejercerían esta acción a través del ácido fólico y la vitamina B12, la cual bloquearía la conversión de la homocisteína en metionina (Fig. 2).
Figura 2. Anticonvulsivantes y homocisteína.
La carbamazepina produce alteraciones hepáticas, que se pueden manifestar como valores anormales en pruebas funcionales, colestasia e ictericia hepatocelular, hepatitis, raramente falla hepática y algunas alteraciones del metabolismo lipídico. Los efectos de la carbamazepina sobre los lípidos sanguíneos y la función hepática son variables y no son consistentes en los diferentes estudios; Se ha descrito aumento del colesterol total y LDL cuyo mecanismo sería la inducción de enzimas microsomales hepáticas, lo que provocaría la alteración del metabolismo de estos lípidos. El fenobarbital y el ácido valproico provocarían las mismas alteraciones. La carbamazepina puede producir también pancreatitis.
El ácido valproico, o ácido 2-propil- pentanoico, es un ácido graso ramificado que produce aumento biológico de amoníaco, bilirrubina, colesterol HDL, testosterona y su proteína transportadora, la globulina de unión a hormonas sexuales; y por otro lado produce disminución biológica de albúmina, testosterona libre, cuerpos cetónicos, colesterol LDL, triglicéridos y de las hormonas prolactina, tirosina y triyodotironina o T3. Alrededor de 80% del ácido valproico se metaboliza directamente en el hígado por glucoronización; el 20% restante se elimina por excreción libre a través de la orina (3%) o a través de la beta oxidación mitocondrial, la misma vía que utilizan los ácidos grasos. Esto significa que el ácido valproico utiliza la misma vía de entrada a la mitocondria que usan los ácidos grasos, la L-carnitina y así se producen los metabolitos inactivos; sin embargo, cuando el ácido valproico captura L-carnitina bloquea su propia metabolización, debiendo recurrir a una vía alternativa que es la metabolización a través del mismo citoplasma, proceso que se conoce como sigma oxidación y que lleva a la producción de ácido 2-propil-4-pentanoico, molécula responsable de la toxicidad del ácido valproico (Fig. 3). El ácido valproico es un medicamento bastante tóxico, por lo que en muchos países se prohíbe su administración en nińos pequeńos.
Figura 3. Metabolización del ácido valproico.
En la beta oxidación, el ácido valproico se une a la coenzima A para penetrar a través de las membranas mitocondriales; posteriormente intercambia coenzima A por carnitina para entrar a la matriz mitocondrial, donde se metaboliza. La captura de carnitina por parte del ácido valproico bloquea parte de la generación de energía de la célula, particularmente la que ocurre durante la metabolización hepática de las grasas; además, la captura de carnitina impide la salida de ésta desde la mitocondria (Fig. 4).
Figura 4. Beta-oxidación del ácido valproico.
El ácido valproico que no logra penetrar utiliza la vía de metabolización de la sigma oxidación, cuyo producto, el ácido-2-propil-4-pentanoico, inhibe a la enzima carbamoil fosfato sintetasa I (CPS I), que es la primera enzima que metaboliza la urea permitiendo la síntesis de urea en el hígado: captura al amoníaco, lo une al bicarbonato y comienza la síntesis de la urea. Este es el mecanismo para detoxificar el nitrógeno excedente del metabolismo de las proteínas; si esto no se produce el resultado es la acumulación de amonio. La falla en la CPS I es la causa de la enfermedad del ciclo de la urea, una enfermedad metabólica grave; el ácido valproico podría generar una falla similar.
Figura 5. Sigma-oxidación del ácido valproico.
El tratamiento con ácido valproico puede producir depleción de carnitina (9), debido a:
La depleción de carnitina tiene los siguientes efectos: reducción del transporte de ácidos grasos de cadena larga y de la beta oxidación de estos ácidos grasos; desvío del metabolismo hacia la sigma oxidación, con producción de ácido-2-propil-4-pentanoico; y acumulación intracelular de amonio en forma similar a lo que ocurre en el déficit de la enzima CPS I. Esto explica los problemas que ocasiona el ácido valproico y porqué se sugiere usar carnitina en aquellos pacientes que usan este fármaco.
Otro problema del ácido valproico es el aumento de peso. Los mecanismos propuestos son múltiples: aumento del apetito, acción directa sobre el hipotálamo, disminución de la termogénesis facultativa y desarrollo de hiperinsulinemia y resistencia a la insulina. En un estudio se observó que los pacientes que utilizaban ácido valproico tenían aumento de insulina y lectina y disminución de grelina y adiponectina, lo que facilitaría el desarrollo de obesidad (10).
La exposición prenatal a fenitoína aumentaría el riesgo de malformaciones congénitas, alteraciones del desarrollo, malformaciones mayores, anomalías menores, anomalías del crecimiento y retraso mental. Estos efectos se pueden observar en una mujer que usa fenitoína, se embaraza y no cambia de anticonvulsivante.
Se ha demostrado que el uso de fenitoína produce una falla en la respuesta de insulina (11). En la Fig. 6 se observa dos tipos de respuesta de insulina frente a una infusión de glucosa de 50 a 300 mg/dL: la respuesta rápida seguida de la respuesta lenta, que ocurre mientras se mantiene la concentración alta de glucosa. La curva control representa la respuesta de individuos normales, que aumentan los niveles de insulina para mantener los niveles de glicemia en rangos normales, mientras que los pacientes que usan fenitoína presentan una respuesta de primera fase muy pequeńa y no presentan respuesta de segunda fase, por lo tanto tienden a desarrollar hiperglicemia. Esto es un problema para pacientes adultos que usan fenitoína y tienen problemas relacionados con el metabolismo de la glucosa, como diabetes o resistencia a la insulina (Fig. 6).
Figura 6. Efecto de aumento de infusión de glucosa (50 a 300 mg/dL) en la secreción de insulina.
Entre los nuevos anticonvulsivantes están topiramato, lamotrigina, levetiracetam, gabapentina, tiagabina y oxicarbazepina.
Topiramato: el más utilizado, tiene como mecanismos de acción: bloquea los canales de sodio dependientes de voltaje en las neuronas; tiene un efecto modulador negativo sobre los receptores de glutamato, que son el medio por el cual producen los cambios anticonvulsivantes; aumenta la actividad de los receptores GABAA; e inhibe la anhidrasa carbónica (isoenzimas II y IV), de ahí sus efectos sobre el estado de los electrolitos. El uso de topiramato se asocia a oligohidrosis, es decir, disminución de la producción de sudor asociada a hipertermia, lo que significa que los pacientes pierden el mecanismo normal de protección frente a hipertermia; esto puede ser beneficioso en los pacientes que sufren de hiperhidrosis, ya que se podría usar como terapia para controlar la sudoración. En los pacientes pediátricos esto se debería al bloqueo de la anhidrasa carbónica, lo que podría modificar la composición del sudor y alterar el resultado del test de sudor en los pacientes con fibrosis quística. Además, el bloqueo de la anhidrasa carbónica produce acidosis metabólica, hipercloremia sin anion gap y bloqueo de la anhidrasa carbónica a nivel del túbulo renal. El uso persistente de topiramato altera el crecimiento y aumenta el riesgo de litiasis renal, por eso se debe evitar su administración en nińos pequeńos. El topiramato también aumenta la sensibilidad a la insulina debido a una alteración en la fosforilación de la kinasa activada por AMPK, lo que lleva a aumento de la adiponectina (12).
Lamotrigina: su uso como monoterapia está autorizado sólo en mayores de 12 ańos y como terapia combinada, desde los dos ańos. Tiene acción sobre el metabolismo del ácido fólico, ya que inhibe la dihidrofolato reductasa; para disminuir el riesgo de trastornos a este nivel se recomienda suplementar con ácido fólico. Es conveniente además evitar el uso concomitante de otros inhibidores del ácido fólico, como el cotrimoxazol. Su uso en el embarazo puede producir anomalías congénitas debido a la alteración del metabolismo del ácido fólico.
Levetiracetam: la FDA no autoriza el uso de este fármaco en menores de 16 ańos. Ejerce su acción a través de la unión a una proteína de la vesícula sináptica, conocida como SV2A. No se ha descrito efectos nutricionales con su uso, pero lleva poco tiempo en el mercado y en la historia de los fármacos se ha visto que los efectos adversos se pueden observar algunos ańos después de que su uso se ha masificado.
Los anticonvulsivantes fenobarbital, carbamazepina, fenitoína, primidona, oxicarbazepina, lamotrigina, topiramato y felbamate tienen algún grado de interferencia con los anticonceptivos orales, disminuyendo la efectividad de éstos.
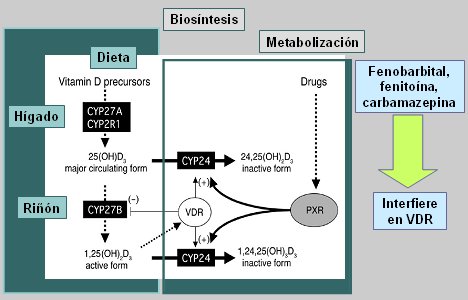 Figura 1. Mecanismo propuesto para desarrollo de osteopenia mediada por PXR (receptor Pregnano X).
Figura 1. Mecanismo propuesto para desarrollo de osteopenia mediada por PXR (receptor Pregnano X).
 Figura 2. Anticonvulsivantes y homocisteína.
Figura 2. Anticonvulsivantes y homocisteína.
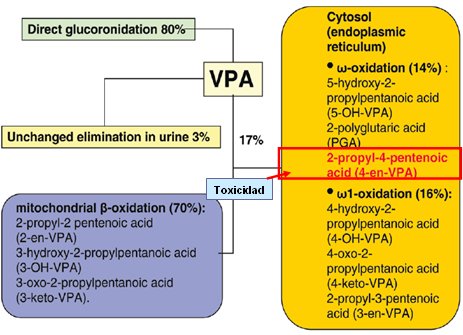 Figura 3. Metabolización del ácido valproico.
Figura 3. Metabolización del ácido valproico.
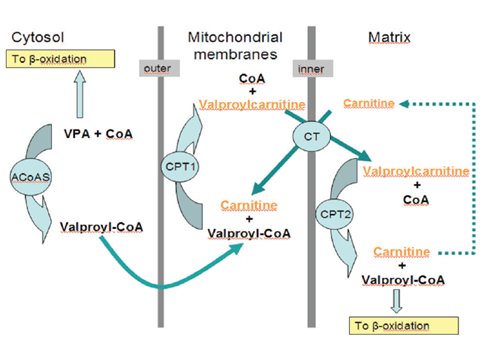 Figura 4. Beta-oxidación del ácido valproico.
Figura 4. Beta-oxidación del ácido valproico.
 Figura 5. Sigma-oxidación del ácido valproico.
Figura 5. Sigma-oxidación del ácido valproico.
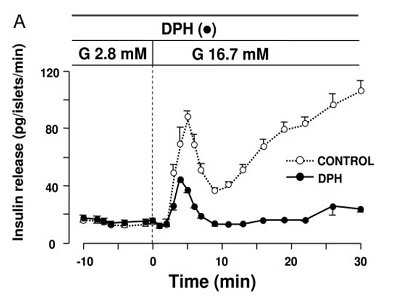 Figura 6. Efecto de aumento de infusión de glucosa (50 a 300 mg/dL) en la secreción de insulina.
Figura 6. Efecto de aumento de infusión de glucosa (50 a 300 mg/dL) en la secreción de insulina.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el marco de las reuniones clínicas de la Unidad General de Cuidados del Nińo del Hospital Padre Hurtado. La publicación de estas actas científicas ha sido posible gracias a una colaboración editorial entre Medwave y la Unidad. El jefe de la UGCN es el Dr. Alejandro Donoso y el Encargado de las Reuniones Clínicas es el Dr. Mario Vildoso.
Autor:
Mario Vildoso Fernández[1]
Citación: Vildoso M. Nutritional effects of anticonvulsants. Medwave 2009 Abr;9(4):e3857 doi: 10.5867/medwave.2009.04.3857
Fecha de publicación: 1/4/2009
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Pascussi JM, Robert A, Nguyen M, Walrant-Debray O, Garabedian M, Martin P, et al. Possible involvement of pregnane X receptor-enhanced CYP24 expression in drug-induced osteomalacia. J Clin Invest. 2005 Jan;115(1):177-86. | PubMed | PMC |Andress DL, Ozuna J, Tirschwell D, Grande L, Johnson M, Jacobson AF, et al. Antiepileptic drug-induced bone loss in young male patients who have seizures. Arch Neurol. 2002 May;59(5):781-6. | CrossRef | PubMed |Burt R, Freston JW, Tolman KG. The influence of phenobarbital on biotransformation of 25-hydroxycholecalciferol. J Clin Pharmacol. 1976 Aug-Sep;16(8-9):393-8. | PubMed |O'Hare JA, Duggan B, O'Driscoll D, Callaghan N. Biochemical evidence for osteomalacia with carbamazepine therapy. Acta Neurol Scand. 1980 Nov;62(5):282-6. | CrossRef | PubMed |Karaaslan Y, Haznedarođlu S, Oztürk M. Osteomalacia associated with carbamazepine/valproate. Ann Pharmacother. 2000 Feb;34(2):264-5. | CrossRef | PubMed |Ranganathan LN, Ramaratnam S. Rapid versus slow withdrawal of antiepileptic drugs. Cochrane Database Syst Rev. 2006 Apr 19;(2):CD005003. | PubMed |Christiansen C, Rodbro P, Lund M. Incidence of anticonvulsant osteomalacia and effect of vitamin D: controlled therapeutic trial. Br Med J. 1973 Dec 22;4(5894):695-701. | CrossRef | PubMed | PMC |Christiansen C, Rodbro P, Nielsen CT. Iatrogenic osteomalacia in epileptic children. A controlled therapeutic trial. Acta Paediatr Scand. 1975 Mar;64(2):219-24. | PubMed |Lheureux PE, Penaloza A, Zahir S, Gris M. Science review: carnitine in the treatment of valproic acid-induced toxicity - what is the evidence? Crit Care. 2005 Oct 5;9(5):431-40. Epub 2005 Jun 10. | CrossRef | PubMed | PMC |Greco R, Latini G, Chiarelli F, Iannetti P, Verrotti A. Leptin, ghrelin, and adiponectin in epileptic patients treated with valproic acid. Neurology. 2005 Dec 13;65(11):1808-9. | CrossRef | PubMed |






 Estudios originales
Estudios originales