Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el marco de las reuniones clínicas de la Clínica Psiquiátrica Universidad de Chile. La publicación de estas actas científicas ha sido posible gracias a una colaboración editorial entre Medwave y la Clínica Psiquiátrica de la Universidad de Chile, cuya directora es la Dra. Graciela Rojas.
La enfermedad de Alzheimer (EA) es una enfermedad neurodegenerativa del cerebro. En el ańo 1995 se pensaba que la EA se producía como consecuencia de un ataque de las proteínas contra las células del cerebro: algo le ocurría a las proteínas que de pronto atacaban y deterioraban la función cerebral. La información disponible en torno a esta enfermedad ha aumentado en forma exponencial en los últimos cinco ańos, después de una época de desarrollo muy lento del conocimiento sobre el tema, probablemente porque la comunidad médica y la industria farmacéutica se abocaron a la búsqueda de elementos terapéuticos sobre la base en una hipótesis equivocada, tal como ocurrió en 1984 con la hipótesis del amiloide, que motivó mucha investigación hasta que se demostró que era errónea.
La proteína tau, que forma parte de los microtúbulos, es fundamental para que éstos permanezcan estables en la neurona; hoy se sabe que en la EA se altera la proteína tau y como consecuencia, los microtúbulos se desarticulan, lo que se correlaciona con cambios intensos, principalmente a nivel del hipocampo, que alteran los fenómenos de plasticidad neuronal, memoria y aprendizaje. Por lo tanto, más que hablar de muerte neuronal en la EA se debe hablar de disfunción neuronal. En las etapas avanzadas de la enfermedad se produce muerte masiva de neuronas. De acuerdo con lo anterior, se puede plantear que la EA es una enfermedad reversible en su etapa inicial.
La EA ataca a las células nerviosas en varias regiones del cerebro; sin embargo, un concepto muy importante es que el primer elemento que se compromete es la glía, es decir, la microglía y los astrocitos y posteriormente se produce el problema degenerativo a nivel neuronal. La EA involucra primero áreas del hipocampo, que es esencial para la memoria de almacenamiento, y algunos núcleos basales compuestos por gran número de neuronas que contienen acetilcolina, sustancia química importante en la memoria y el aprendizaje; en etapas más avanzadas la enfermedad compromete la corteza cerebral, que participa en el pensamiento consciente y el lenguaje. Por ello, los enfermos con EA presentan alteraciones importantes de la capacidad de lenguaje y en una etapa avanzada no pueden expresar ideas, sino sólo sonidos guturales.
A nivel histológico, lo primero que se observa en la EA es la aparición de ovillos neurofibrilares y placas seniles, formadas por deposición del polímero beta amiloide. Las placas seniles se pueden encontrar en grandes cantidades en individuos seniles sin EA, mientras que los ovillos neurofibrilares son patognomónicos de esta enfermedad. Inicialmente no se observa pérdida neuronal, pero a medida que pasa el tiempo esta pérdida avanza rápidamente en comparación con el proceso de envejecimiento normal, en el cual se pierde hasta 10% de las neuronas; en la EA la pérdida es mucho mayor y la curva de decaimiento es muy acentuada en las últimas etapas (Fig. 1).
Figura 1. Pérdida neuronal progresiva en la enfermedad de Alzheimer.
La EA depende de factores ambientales y genéticos. La variante de tipo familiar, que corresponde a 2% de los casos, se debe a mutaciones en tres tipos de genes: las presenilinas, la proteína precursora del amiloide y la IL-1, que causan una enfermedad de aparición precoz, alrededor de los 35 a 45 ańos de edad. La EA de tipo esporádico, que representa 98% de los casos, se explica por la presencia de genes de susceptibilidad que hacen que la persona sea vulnerable a desarrollar la enfermedad a una edad más avanzada. El más conocido de estos genes es el de la apolipoproteína E, que posee cuatro alelos; el aumento del alelo 4 es un factor de alto riesgo para padecer EA. Por otra parte, se ha determinado claramente la importancia de ciertos factores ambientales en el desarrollo de la EA y hoy se sabe que el cambio en una serie de elementos del estilo de vida previene su desarrollo.
El envejecimiento cerebral aumenta la vulnerabilidad debido a que se asocia a: disminución de la capacidad de regeneración neuronal y de las sinapsis; agregación proteica; alteraciones del citoesqueleto; dańo por radicales libres; disminución de factores tróficos y neuroinflamación. La falta de trofismo impide la adecuada arborización y formación de nuevos botones sinápticos, hechos claves para la plasticidad y el aprendizaje. Por otra parte, existen agregaciones proteicas o polímeros que pueden ser normales o anormales; los polímeros anormales están conformados por la proteína tau.
El envejecimiento es un proceso natural en el que participa una combinación de elementos ambientales y el genoma. Este proceso se puede retardar si durante la vida del individuo se promueven los siguientes aspectos:
Numerosos trabajos han demostrado que el estrés causa envejecimiento celular. Los cromosomas poseen en su región terminal ciertas secuencias de material genético que se acortan a medida que aumenta la edad: son los telómeros y la enzima que los repara es la telomerasa. El acortamiento de los telómeros hace que la célula vaya perdiendo la capacidad de vivir y que los tejidos vayan reduciendo su funcionalidad. Se ha demostrado que en las personas sometidas a estrés crónico se producen alteraciones en la actividad de las telomerasas y por lo tanto, en su capacidad para reparar los telómeros.
El estrés se produce cuando la persona percibe determinado cambio que le genera incertidumbre y le hace sentir que existe riesgo de una amenaza. La globalización, los cambios económicos, la aparición de nuevas tecnologías y cambios en el medio ambiente, la digitalización, la erosión de los valores tradicionales y el aumento de las expectativas de vida son situaciones que predisponen al estrés. Se ha demostrado que existe una relación directa entre estrés crónico y neurodegeneración, en términos de pérdida del número de conexiones sinápticas. Por el contrario, la práctica de la meditación facilita el establecimiento de conexiones sinápticas y circuitos neuronales en ciertos dominios del cerebro, como el hipocampo, lo que facilitaría los fenómenos de plasticidad.
En noviembre de 2007 se realizó una Conferencia Internacional titulada “Hipótesis actuales sobre la EA”, en la Facultad de Medicina de la Universidad de Chile. El evento reunió a expertos de todo el mundo y a especialistas chilenos para analizar el panorama actual de la enfermedad, tanto en lo que se refiere a su patogenia como a las nuevas avenidas terapéuticas. En la página web de la Alzheimer’s Association aparece en forma destacada la hypotesis factory, es decir, la fábrica de hipótesis, lo que refleja la gran cantidad de controversias y discusión que suscita el tema. Es por ello que se ha creado el grupo de investigación del Centro Internacional de Biomedicina (ICC), un grupo básico clínico que involucra a psicólogos, neurólogos y psiquiatras, dedicado al estudio de la EA.
La hipótesis de la cascada de amiloide plantea que en la superficie de las neuronas existe una proteína denominada proteína precursora del amiloide (APP), la cual es cortada por las enzimas secretasas alfa, beta y gam, generando el péptido beta amiloide (A/beta), que la célula primero internaliza y luego externaliza, depositándose en el extracelular. Ese péptido por sí solo no produce ningún dańo, pero a medida que se acumula se empieza a autoagregar, formando pequeńos agregados de oligómeros de 6 a 8 unidades de Abeta, de 4 Kd de peso y 40 aminoácidos. Este agregado de varios Abeta es lo peligroso y no la placa senil, como se pensó durante mucho tiempo (Fig. 2).
Figura 2. La hipótesis de la cascada de amiloide.
Un elemento importante en la comprensión de la EA es la proteína tau, descubierta por nuestro grupo de investigación en 1974, en la Universidad de Colorado, en el curso del análisis del citoesqueleto neuronal(Fig. 3).
Figura 3. Formación de placas seniles en la EA.
Lo primero que se determinó fue que la proteína tau es responsable de la estabilización de los microtúbulos; posteriormente varios laboratorios, incluyendo el nuestro, definieron el papel de dicha proteína en la formación de los filamentos pareados helicoidales involucrados en la EA. La hipótesis de tau estableció que esta proteína forma los ovillos neurofibrilares (NFT), tan raros de encontrar en sujetos normales que se consideran como un signo patognomónico de la EA y de las taupatías y otras enfermedades relacionadas, como la demencia frontotemporal del cromosoma 17, en la cual la mutación del gen de la proteína tau de dicho cromosoma se asocia a demencia precoz, de aparición entre los 35 y 40 ańos de edad. Esta demencia no es una EA, porque no hay formación de placas ni otras estructuras, sino solamente los agregados de la proteína tau; esta proteína normalmente forma parte del citoesqueleto, pero en este caso pierde su estructura y forma polímeros anómalos, los filamentos pareados helicoidales (Fig. 4).
Figura 4. La hipótesis de tau y los ovillos neurofibrilares (NFT).
La EA es una enfermedad multifactorial; la alteración de tau constituye una vía común para la mayoría de las seńales anómalas causadas por la degeneración neuronal. Entre los factores involucrados en la génesis de la EA están los siguientes: compromiso de la proteína tau nuclear; estrés oxidativo; alteraciones en la respuesta inmune; activación de células gliales, con producción de citoquinas proinflamatorias y seńales de crecimiento anómalas; anormalidades proteicas; infecciones virales; alteraciones sinápticas, aluminio y otras toxinas, etc. Todas las alteraciones de los mecanismos de seńalización que llegan al interior de la neurona se producen como consecuencia de la fosforilación de la proteína tau; si no hay fosforilación, no hay EA. Por lo tanto, la EA se puede producir por diferentes factores, pero el paso final común de todos ellos es la fosforilación de tau, paso clave para que se formen los ovillos neurofibrilares. Las taupatías son enfermedades causadas por la formación de polímeros de la tau que forman los ovillos neurofibrilares, los que conducen al desarrollo de la demencia tipo frontotemporal del cromosoma 17 u otras enfermedades neurodegenerativas (Fig. 5).
Figura 5. Patogenia de las taupatías. PHF = polímeros de la proteína tau.
Un elemento que apoya fuertemente la hipótesis de tau es que si se cambia la secuencia de aminoácidos de esta proteína o si se produce alguna mutación en ella, como ocurre en las demencias frontotemporales, la proteína tau no actúa estabilizando el citoesqueleto neuronal y se forman polímeros autónomos, lo que resulta en muerte neuronal masiva (Fig. 6).
Las fosforilaciones más precoces de tau que se han detectado en la EA son las de phosphoserine 202, en las regiones CA1 y CA2 del hipocampo, donde se privilegian todos los procesos sinápticos. Esta alteración se traduce en la presencia de abundantes ovillos neurofibrilares, lo que apoya la hipótesis del rol de la proteína tau (Fig. 7).
Los conocimientos actuales sobre la EA han permitido desterrar varios mitos. Hoy se sabe que:
La hipótesis de la neuro-inmuno-modulación, planteada por nuestro grupo, es la base del enfoque terapéutico moderno de la EA. Dicha hipótesis plantea que en la EA hay un cambio en la inmunidad innata, es decir, en los procesos más inespecíficos del sistema inmune. Hay una serie de seńales de peligro (danger signals), entre las cuales está el colesterol elevado, la glicemia sobre 120 mg/dl de manera sostenida durante mucho tiempo y la resistencia a la insulina, que conducen a la producción de partículas de LDL oxidadas que, al igual que los oligómeros del amiloide o la proteína S100, afectan a los receptores TLR y RAGE. Estos receptores se encuentran en la superficie de la microglía, que por lo tanto es el primer tejido afectado por las seńales externas anómalas; como resultado la microglía se sobreactiva, produciendo cantidades exageradas del factor de transcripción NF-kappa-beta, que normalmente regula la expresión de los genes. En este caso, dicho factor aumenta la liberación de TNF-alfa, una citoquina pro-inflamatoria que a su vez estimula la producción de IL-1 e IL-6. La mayor producción de agentes pro-inflamatorios produce seńales anómalas a nivel de diversos receptores neuronales, ocasionando una reacción neuronal anómala que se manifiesta por la activación de ciertos sistemas enzimáticos (Fig. 8).
Figura 8. Hipótesis de la neuro-inmuno-modulación en la enfermedad de Alzheimer.
En el ańo 1997 nuestro grupo descubrió que lo primero que ocurre en la EA es la activación de la enzima Cdk5, una quinasa que regula el ciclo celular y que en el cerebro desempeńa una importante función relacionada con el desarrollo y la neurogénesis. La Cdk5 aumenta alrededor de diez veces e hiperfosforila a la proteína tau, la que como consecuencia forma agregados que llevan a la muerte neuronal. Por lo tanto, la cadena patogénica parte con las alteraciones de la inmunidad innata que se genera a nivel de la glía, la cual envía seńales erróneas que son interpretadas por la neurona de tal forma que producen dańo neuronal y neurodegeneración. Esta es la hipótesis unificada, llamada así porque abarca a casi todas las otras hipótesis planteadas anteriormente. En la siguiente imagen se muestra un esquema de la gran unidad neural: neurona-glía-vasos; una neurona glutamaérgica recibe seńales de la glía, en forma de metabolitos que necesita para realizar su actividad sináptica; por otro lado se libera una serie de factores tróficos y marcadores pro-inflamatorios que llegarán al interior de la neurona, mientras que los vasos sanguíneos suministran la glucosa y metabolitos que requieren la glía y la neurona (Fig. 9).
Figura 9. La gran unidad neural: neurona-glía-vasos.
A continuación se analizará los mayores problemas que se deben resolver para avanzar en la comprensión de la patogenia de la EA, sobre la base de que se trata de una enfermedad multifactorial.
Cómo Abeta afecta el comportamiento de tau y su fosforilación: En los ańos 90, cuando se plantearon las hipótesis del amiloide y de la proteína tau, para conectar ambos elementos nuestro grupo hizo estudios en un modelo experimental de cerebros embrionarios de ratas de 18 días en los cuales se disecaba el hipocampo para luego cultivar las células. Al analizar el comportamiento del cultivo de células del hipocampo frente al agregado de amiloide se encontró que la mayor parte de las neuronas había desaparecido a las 72 horas. Si al mismo cultivo se agregaba amiloide y butirolactona, molécula que controla a la enzima Cdk5, no moría ninguna neurona, con lo que se comprobó la hipótesis del rol de la sobreactivación de la enzima Cdk5 en la etapa subclínica de la EA, la cual planteaba que los oligómeros del amiloide afectan a receptores que activan a la proteína P35, que se asocia a la Cdk5 haciéndola más activa; luego, ese complejo más activo fosforila a la tau. Esto orientó la búsqueda de nuevas terapias hacia la producción de fármacos y moléculas dirigidas al control de la enzima Cdk5 (Fig. 10).
En la siguiente imagen se observa el modelo molecular que nuestro grupo desarrolló en el laboratorio mediante técnicas computacionales, después de cristalizar la enzima y someterla a difracción de rayos X. Este modelo permite aplicar drogas y determinar cuál de ellas tiene un efecto inhibitorio sobre la enzima.
Figura 11. Modelamiento molecular de la enzima Cdk-5, con su regulador p35.
La relación entre amiloide y patología en modelos animales: Esto se comprobó mediante técnicas de inmunocitoquímica en el modelo transgénico de EA Tg 2576, en el cual se desarrolla este mismo tipo de agregados, es decir, placas seniles, pero también hay acúmulos de la proteína tau alterada en el cerebro de los animales transgénicos, en comparación con el cerebro normal. Por lo tanto, se puede reproducir en modelos transgénicos lo mismo que se observa en el cerebro humano (Fig. 12).
Figura 12. Inmunohistoquímica de epítopes P-tau.
La proteína tau se fosforila en tirosina, además de las fosforilaciones en serina y treonina. Este fenómeno, descubierto recientemente, es propio de la familia de quinasas Src, que son proteínas oncogénicas de la superficie neuronal que aparentemente juegan un rol en la EA.
El compromiso de la proteína tau nuclear en el cerebro de pacientes con EA está comprobado. La proteína se localiza en las regiones nucleolares de la neurona junto con el marcador NOR-90. Esta proteína tau nuclear jugaría un rol en la EA en asociación con la proteína Pin 1, peptidil-prolil cis-trans isomerasa o hPPIasa, que es protectora, ya que cambia la conformación de la tau evitando que se hiperfosforile. Cuando Pin 1 disminuye deja a la proteína tau a merced de las quinasas, especialmente de la Cdk5, que procede a realizar la hiperfosforilación (Fig. 13).
El estrés oxidativo cambia el patrón de las quinasas a favor de una fosforilación anómala, lo cual también ha sido demostrado por nuestro grupo en el laboratorio; se activa la Cdk5 con las consecuencias ya descritas, aumentando la formación de filamentos pareados helicoidales de la proteína tau (Fig. 14).
Figura 14. El estrés oxidativo favorece la formación de ovillos neurofibrilares.
La inflamación tiene un rol importante en la degeneración neuronal, ya que es un proceso determinado por una serie de factores moleculares que generan proteínas de fase aguda, proliferación y diferenciación. Las citoquinas son importantes en la regulación de ciertos procesos a nivel de proliferación celular y control de la diferenciación, en las cantidades que normalmente libera la glía; sin embargo, cuando los niveles de citoquinas suben cien veces o más, como ocurre en la EA, la situación pasa al otro extremo, porque en la neurona hay ciertos receptores que no reconocen a las citoquinas hasta que los niveles de éstas están muy elevados: son los llamados receptores de muerte, como el P75, que empieza a reconocer a estas citoquinas anómalas y envía seńales para que se produzca la apoptosis. En la siguiente imagen se muestra el modelo que desarrolló nuestro grupo para describir el rol de la neuroinflamación en la patogénesis de la EA, modelo que ha sido la base para el uso de drogas antiinflamatorias en el tratamiento de esta enfermedad. El ibuprofeno y la indometacina actúan actúan a nivel de los ligandos del PPAR, los cuales impiden la producción de TNF-α y de IL-6 e interactúan con ciertos receptores en la neurona, aumentando la Cdk5 y la fosforilación de la proteína tau.
Figura 15. Modelo que describe el rol de los PPAR y la neuroinflamación en la patogénesis de la EA.
La alteración de los mecanismos de seńalización es importante en el desarrollo de la EA. En esto participan el estrés oxidativo y factores como las citoquinas, que actúan por la vía de los receptores NMDA o de receptores para las citoquinas activando la vía JAKs/STATs y aumentando la producción de p38, para posteriormente activar un factor de transcripción que va a elevar la producción del gen de la p35. Ésta activa a la Cdk5, lo que aumenta la fosforilaración de la proteína tau. En este proceso está involucrado el calcio intracelular: el aumento de la concentración de calcio intracelular facilita los procesos que conducen a la neurodegeneración (Fig. 16).
Figura 16. Alteración de los mecanismos de seńalización en la patogenia de la EA.
Los astrocitos activados liberan óxido nítrico, IL-1 beta, TNF-alfa y NGF, que es crítico para que se desarrollen los procesos sinápticos y la plasticidad neuronal en el cerebro adulto. La glía, activada por todos los factores de dańo seńalados, aumenta excesivamente la producción de NGF, el cual además se procesa en forma anormal, de modo que llega hasta a la neurona donde activa al factor P75, que es un factor de muerte neuronal, favoreciendo la neurodegeneración. El NGF en exceso puede ser altamente dańino (Fig. 17).
Figura 17. Degeneración, disfunción neuronal y muerte neuronal.
Los receptores colinérgicos también están involucrados. Se ha demostrado que ciertos agonistas nicotínicos protegen a la neurona contra los efectos de las seńales anómalas; por lo tanto los receptores nicotínicos, principalmente los alfa 7, tienen una acción protectora contra la neurodegeneración. Esta es una de las razonas de porqué la nicotina hace que la persona sea menos proclive a desarrollar la EA. Asimismo, se ha demostrado que la actina del citoesqueleto se desorganiza cuando actúan ciertas seńales activadas por la vía patogénica Rac (9). En abril de 2008, nuestro grupo descubrió que la acumulación de hierro en la sangre es un detonante del estrés oxidativo que conduce a la EA. El ser humano acumula hierro durante toda su vida, porque este mineral no se elimina. En nuestro estudio se demostró que había alteraciones importantes en los niveles de hierro libre redox (Fe+3), que tiene un alto potencial oxidativo, en el líquido cefalorraquídeo de pacientes con EA en comparación con los controles.
La EA se diagnostica con seguridad sólo después de la muerte. El diagnóstico en vida tiene 85% de certeza, porque se puede confundir con un trastorno cognitivo leve. Por esto es importante desarrollar herramientas para lograr un diagnóstico certero y precoz durante la vida del paciente, entendiendo como precoz la etapa subclínica, es decir, diez a veinte ańos antes de que la persona tenga manifestaciones de EA. Para lograr este objetivo se están abriendo dos avenidas: los biomarcadores moleculares y las neuroimágenes.
Las neuroimágenes, aún con todas las variantes tecnológicas disponibles, no tienen mayor utilidad en este momento, ya que ni el especialista más experto puede distinguir un cerebro normal de un cerebro con EA; pero en este momento nuestro grupo está trabajando en la tecnología PET de neuroimágenes, con la cual se ha logrado identificar los ovillos neurofibrilares con radiomarcadores. Alrededor de quinientos grupos de trabajo intentaron aplicar esta tecnología en el estudio de la EA, pero no tuvieron buenos resultados porque todos trataron de identificar las placas seniles en el cerebro, con base en una hipótesis errada. El trabajo de Klunk, publicado en la revista Nature, fue el más citado en toda la historia de la ciencia, pero el compuesto que se utilizó no tuvo beneficios a nivel clínico a pesar de todo el dinero que se gastó.
Nuestro grupo de investigación ha generado una tecnología de neuroimágenes con base en un trazador específico para EA. Este trazador forma parte de una familia de moléculas inocuas que se utilizan para otros problemas en Medicina y se fijan a los ovillos neurofibrilares en el cerebro. En el ańo en curso, 2008, la aplicación de esta tecnología se encuentra en etapa de investigación clínica. Una de las moléculas que se utiliza es lanzoprazol, que se fija a los ovillos neurofibrilares y permite un diagnóstico patognomónico de estas estructuras. En un estudio longitudinal de siete ańos de duración se analizó una muestra de LCR de pacientes con EA con trastorno cognitivo leve y normales y se encontró una estrecha relación entre el deterioro cognitivo, medido por todos los test, y los niveles de proteína tau fosforilada. Los niveles de tau hiperfosforilada aumentaban en la misma proporción que el deterioro cognitivo, con una curva prácticamente lineal, lo que sugiere que la medición de estos niveles en el LCR es un excelente medio para detectar precozmente la EA. Actualmente hay kits que se utilizan con dicho fin.
Otro test que está en desarrollo se basa en una muestra de sangre en la cual se separan las plaquetas, elementos que poseen varios marcadores del sistema nervioso central, entre ellos una proteína tau pesada, anómala, que es exclusiva de los pacientes con EA. El patrón electrofóretico, a través de un algoritmo matemático, permite identificar si una persona tiene EA y determinar su grado de deterioro cognitivo.
Las intervenciones a nivel de las placas de amiloide no tienen resultados; es posible actuar a nivel de las enzimas que fosforilan a la proteína tau y hay todo un campo nuevo en relación con los antiinflamatorios.
La nanotecnología, en el futuro, permitirá encapsular drogas en micelas o en oro coloidal para hacerlas llegar hasta el cerebro; gracias a estos nanobots se podrá observar las alteraciones del cerebro humano, efectuar un diagnóstico precoz y realizar cirugías moleculares.
También se deberá desarrollar la terapia con base en células madres; de hecho no sería necesario utilizar estas células, sino que bastaría con activar los circuitos que están en desuso a partir de células madres que ya existen en el cerebro. Los avances en esta área aún son lentos.
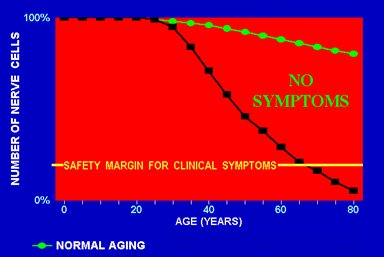 Figura 1. Pérdida neuronal progresiva en la enfermedad de Alzheimer.
Figura 1. Pérdida neuronal progresiva en la enfermedad de Alzheimer.
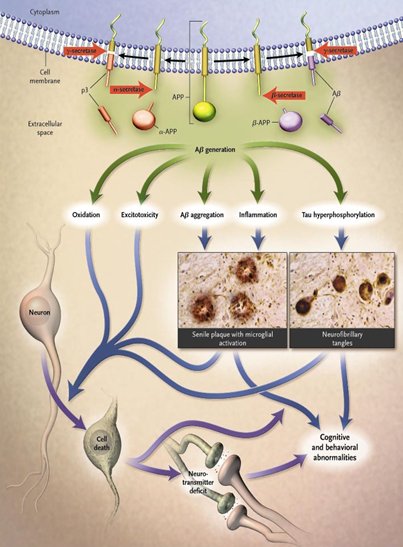 Figura 2. La hipótesis de la cascada de amiloide.
Figura 2. La hipótesis de la cascada de amiloide.
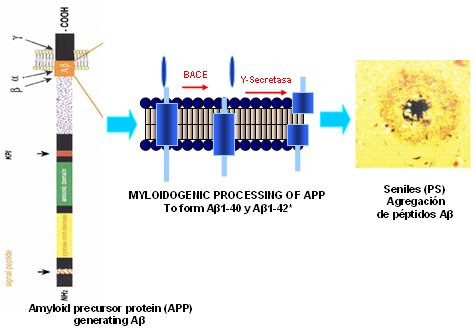 Figura 3. Formación de placas seniles en la EA.
Figura 3. Formación de placas seniles en la EA.
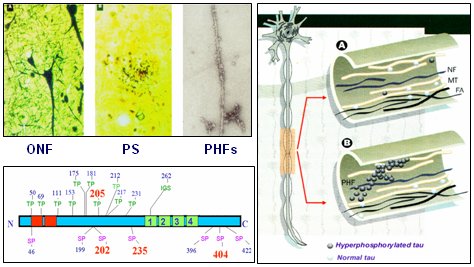 Figura 4. La hipótesis de tau y los ovillos neurofibrilares (NFT).
Figura 4. La hipótesis de tau y los ovillos neurofibrilares (NFT).
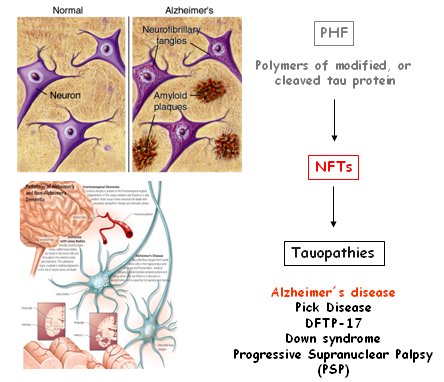 Figura 5. Patogenia de las taupatías. PHF = polímeros de la proteína tau.
Figura 5. Patogenia de las taupatías. PHF = polímeros de la proteína tau.
 Figura 6. El cambio en un solo aminoácido (P301L) de la proteína tau resulta en muerte neuronal masiva, con acumulación de tau.
Figura 6. El cambio en un solo aminoácido (P301L) de la proteína tau resulta en muerte neuronal masiva, con acumulación de tau.
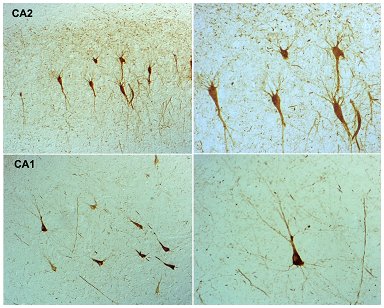 Figura 7. Fosforilaciones precoces de tau en las regiones CA1 y CA2 del hipocampo en la enfermedad de Alzheimer: phosphoserine 202.
Figura 7. Fosforilaciones precoces de tau en las regiones CA1 y CA2 del hipocampo en la enfermedad de Alzheimer: phosphoserine 202.
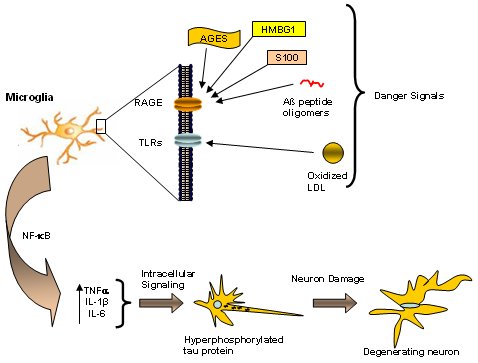 Figura 8. Hipótesis de la neuro-inmuno-modulación en la enfermedad de Alzheimer.
Figura 8. Hipótesis de la neuro-inmuno-modulación en la enfermedad de Alzheimer.
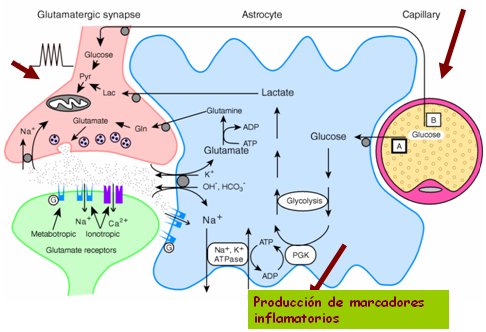 Figura 9. La gran unidad neural: neurona-glía-vasos.
Figura 9. La gran unidad neural: neurona-glía-vasos.
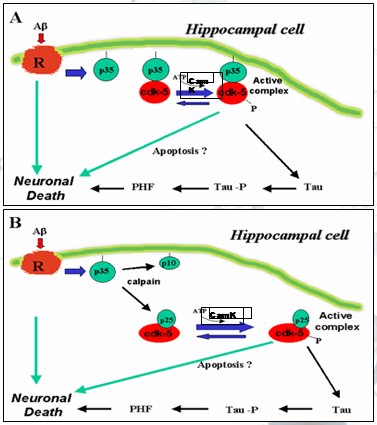 Figura 10. La pérdida de la regulación de la enzima Cdk5 es crítica en la degeneración neuronal que conduce a la EA.
Figura 10. La pérdida de la regulación de la enzima Cdk5 es crítica en la degeneración neuronal que conduce a la EA.
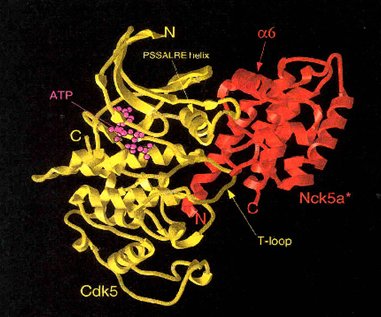 Figura 11. Modelamiento molecular de la enzima Cdk-5, con su regulador p35.
Figura 11. Modelamiento molecular de la enzima Cdk-5, con su regulador p35.
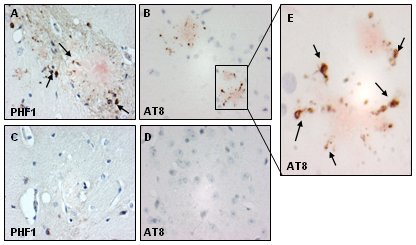 Figura 12. Inmunohistoquímica de epítopes P-tau.
Figura 12. Inmunohistoquímica de epítopes P-tau.
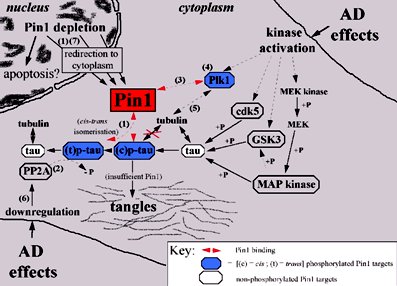 Figura 13. Pin1 restaura la actividad de unión a microtúbulos de tau a través de la interacción directa del dominio WW de Pin1 con la región rica en prolinas de tau.
Figura 13. Pin1 restaura la actividad de unión a microtúbulos de tau a través de la interacción directa del dominio WW de Pin1 con la región rica en prolinas de tau.
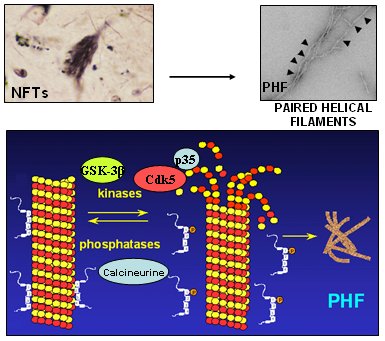 Figura 14. El estrés oxidativo favorece la formación de ovillos neurofibrilares.
Figura 14. El estrés oxidativo favorece la formación de ovillos neurofibrilares.
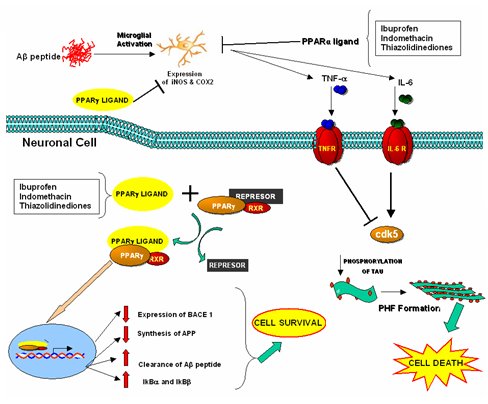 Figura 15. Modelo que describe el rol de los PPAR y la neuroinflamación en la patogénesis de la EA.
Figura 15. Modelo que describe el rol de los PPAR y la neuroinflamación en la patogénesis de la EA.
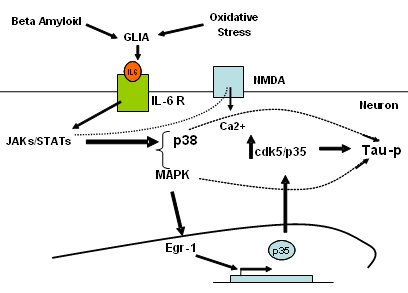 Figura 16. Alteración de los mecanismos de seńalización en la patogenia de la EA.
Figura 16. Alteración de los mecanismos de seńalización en la patogenia de la EA.
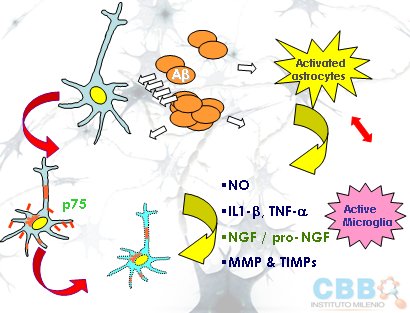 Figura 17. Degeneración, disfunción neuronal y muerte neuronal.
Figura 17. Degeneración, disfunción neuronal y muerte neuronal.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el marco de las reuniones clínicas de la Clínica Psiquiátrica Universidad de Chile. La publicación de estas actas científicas ha sido posible gracias a una colaboración editorial entre Medwave y la Clínica Psiquiátrica de la Universidad de Chile, cuya directora es la Dra. Graciela Rojas.
Expositor:
Ricardo Maccioni Baraona[1]
Citación: Maccioni R. New avenues toward the diagnosis and treatment of cognitive disorders: Alzheimer's disease. Medwave 2008 Dic;8(11):e3660 doi: 10.5867/medwave.2008.11.3660
Fecha de publicación: 1/12/2008
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, et al, Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004 Dec 7;101(49):17312-5. Epub 2004 Dec 1. | CrossRef | PubMed | PMC |Maccioni RB, Muńoz JP, Barbeito L. The molecular bases of Alzheimer's disease and other neurodegenerative disorders. Arch Med Res. 2001 Sep-Oct;32(5):367-81. | CrossRef | PubMed |Fernández JA, Rojo L, Kuljis RO, Maccioni RB. The damage signals hypothesis of Alzheimer's disease pathogenesis. J Alzheimers Dis. 2008 Jul;14(3):329-33. | PubMed |Maccioni RB, Otth C, Concha II, Muńoz JP. The protein kinase Cdk5. Structural aspects, roles in neurogenesis and involvement in Alzheimer's pathology. Eur J Biochem. 2001 Mar;268(6):1518-27. | CrossRef | PubMed |Mendoza-Naranjo A, Gonzalez-Billault C, Maccioni RB. Abeta1-42 stimulates actin polymerization in hippocampal neurons through Rac1 and Cdc42 Rho GTPases. J Cell Sci. 2007 Jan 15;120(Pt 2):279-88. Epub 2007 Jan 2. | CrossRef | PubMed |Lavados M, Guillón M, Mujica MC, Rojo LE, Fuentes P, Maccioni RB. Mild cognitive impairment and Alzheimer patients display different levels of redox-active CSF iron. J Alzheimers Dis. 2008 Mar;13(2):225-32. 






 Estudios originales
Estudios originales