Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Palabras clave: congenital anomalies, morphological development, malformations, deformations, disruptions, dysplasias
Las anomalías congénitas en conjunto con la restricción del crecimiento intrauterino dan cuenta del 50 a 60% de la mortalidad fetal. En este artículo se describen las malformaciones congénitas más importantes divididas por sistemas, la mayoría son diagnosticables a través del ultrasonido; sin embargo, hay un grupo de ellas que no son posibles de diagnosticar, otras que se desarrollan tardíamente en el embarazo y finalmente existen casos en que el examen ultrasonográfico puede sugerir una anormalidad inexistente. También se profundiza en la incidencia, etiología y factores de riesgo de las malformaciones congénitas, se describe la importancia y características del diagnóstico prenatal y de la prevención por medio de la suplementación de ácido fólico en la dieta.
Las anomalías congénitas constituyen un tema de importancia en obstetricia, tanto por su pronóstico vital, ya que en conjunto con la restricción del crecimiento intrauterino dan cuenta del 50 a 60 % de la mortalidad fetal; como por el desarrollo que ha tenido su diagnóstico prenatal.
A continuación se describen las malformaciones congénitas más importantes divididas por sistemas; la mayoría son diagnosticables a través del ultrasonido; sin embargo, hay un grupo de ellas que no son posibles de diagnosticar, otras que se desarrollan tardíamente en el embarazo (después de las 24 semanas); y, finalmente, existen casos en que el examen ultrasonográfico puede sugerir una anormalidad inexistente.
Se entiende como anomalía congénita cualquier trastorno del desarrollo morfológico, estructural o funcional de un órgano o sistema presente al nacer. Puede ser familiar o esporádica, externa o interna, y única o múltiple. Según su origen se subdividen en malformaciones, deformaciones, disrupciones y displasias.
Algunas definiciones básicas se describen a continuación:
Finalmente, las anomalías congénitas se clasifican como mayores o menores, entendiéndose como anomalía congénita mayor la que representa un riesgo vital, requiere de cirugía o implica secuelas estéticas severas, y menor si no presenta secuelas estéticas significativas, ni alteraciones en la calidad o esperanza de vida del paciente1,2.
Las anomalías congénitas tienen una incidencia de alrededor del 5% en forma global, siendo las anomalías congénitas mayores entre un 1,8 y 3%, presentándose en uno de cada 30 recién nacidos vivos y en 0,1 a 1 de cada 10 mortinatos1,2.
Las anomalías congénitas mayores representan por sí solas el 25% de la mortalidad perinatal, siendo la primera causa de mortalidad infantil en Chile.
En orden de frecuencia, el primer lugar lo comparten, con un 21% del total, las malformaciones cardíacas y genitourinarias, siguiendo en frecuencia con un 16% las del sistema nervioso central y luego las musculoesqueléticas, faciales y gastrointestinales con un 5 a 7% cada una.
En la Tabla I se muestra la tasa de detección de anomalías congénitas por sistemas publicadas en el extranjero y en Chile:
| Sistema | Extranjero % | Chile% |
Cardiovascular SNCCara GastrointestinalOsteomuscular UrogenitalCromosómico Otros | 18 8453 2129 5031 20 | 36 7720 5553 7947 28 |
Tabla I. Tasa de detección de anomalías congénitas.
Las anomalías congénitas tienen diferentes etiologías, las que pueden dividirse en 4 subgrupos ver Tabla II:
| Genéticas | 12-15% (anomalías cromosómicas y mutaciones genéticas) |
| Factores ambientales | 7-10% |
| Multifactoriales | 20-25% |
| Desconocidas | 50-60% |
Tabla II. Etilogías de las anomalías congénitas.
Dentro de nuestro campo de estudio merecen especial mención las aneuploidías, tanto por su valor pronóstico, como por el desarrollo que se ha logrado en el diagnóstico prenatal de éstas3,4.
Se llama aneuploidía al defecto genético dado por una alteración en el número de cromosomas, siendo las más frecuentes las trisomías, y dentro de éstas las trisomías 13, 18 y 21. Cada una de ellas presenta un síndrome característico, con múltiples malformaciones y un fenotipo distintivo al nacer.
Las aneuploidías en globo representan más del 50% de los abortos espontáneos y el 5% de los mortinatos mayores de 28 semanas de edad gestacional, siendo su frecuencia individual:
En la Tabla III se muestran las malformaciones pesquisables por ultrasonido en estas aneuploidías5:
Tabla III. Malformaciones pesquisables por ultrasonido.
La base del diagnóstico prenatal de las anomalías congénitas lo constituye la ecografía obstétrica, en especial la efectuada entre las 18 y 24 semanas de edad gestacional6. En este examen se sugiere hacer una revisión ordenada y sistemática como se describe a continuación:
Sin embargo, además de la ecografía obstétrica se han desarrollado métodos más precoces de detección de anomalías y aneuploidías, en especial en países donde se ha legalizado el aborto7. Por no ser ésta la realidad de América Latina, sólo mencionaremos los de uso más extendido:
Anencefalia
Malformación congénita caracterizada por ausencia total o parcial del cráneo, la piel que lo recubre y la masa encefálica, producida por un defecto en el cierre anterior del tubo neural, se asocia con espina bífida y onfalocele.
Su incidencia varía geográficamente y según la población estudiada, entre 1 en mil nacimientos en Estados Unidos, hasta 1 en 100 en algunas regiones del Reino Unido.
El diagnóstico de anencefalia durante el segundo trimestre se basa en la demostración de ausencia de calota y hemisferios cerebrales. Sin embargo, los huesos faciales, tronco encefálico y porciones de los huesos occipitales y del mesencéfalo están usualmente presentes. En hasta un 50% de los casos se pueden ver lesiones espinales asociadas.
En el primer trimestre, se puede hacer el diagnóstico después de las 11 semanas, cuando normalmente ocurre la osificación del cráneo. Existiría una progresión en el desarrollo del defecto desde acrania, posteriormente a exencefalia y finalmente anencefalia.
La anencefalia es invariablemente letal, aproximadamente en el 50% de estos casos se produce mortalidad intrauterina, y el resto muere dentro de las pocas horas de nacidos.
Encefalocele
Malformación congénita en que existe protrusión del cerebro y/o meninges a través de un defecto de la calota.
Es el resultado de una falla del cierre de la porción craneal del tubo neural durante el primer mes del período embrionario. Se asocia con frecuencia a otras malformaciones, tales como la hidrocefalia, iniencefalia, labio leporino, malformaciones cardíacas y genitourinarias. Más de 30 síndromes descritos incluyen el encefalocele entre sus características.
Tiene una incidencia de 1 en 2 mil nacidos vivos, presentándose en igual proporción en el sexo femenino y masculino.
El diagnóstico se hace a través de la observación de una masa paracraneana, la cual en el 75% de los casos es occipital, en el 12% frontal y en el 13% parietal. El tumor puede estar constituído por líquido, por masa encefálica o por ambos. Si la mayor parte del cerebro se encuentra en el encefalocele, se ve también microcefalia.
El pronóstico depende de la asociación a otras malformaciones cerebrales y del diagnóstico sindromático. El tamaño no es de valor pronóstico, ya que grandes encefaloceles pueden no contener elementos neurales.
Se recomienda, además del estudio ecográfico, realizar un cariograma para descartar trisomía 13 y la realización de una ecocardiografía para descartar las malformaciones cardíacas usualmente presentes en los síndromes que incluyen al encefalocele entre sus características.
La vía de parto recomendada va a depender del tamaño del defecto, la cantidad de masa encefálica herniada y las anomalías asociadas.
Espina bífida y mielomeningocele
La espina bífida corresponde a una familia de defectos en el cierre de la columna vertebral, que se caracterizan por la protrusión o exposición de la médula espinal y/o las meninges a través del defecto mencionado.
Este grupo de defectos se pueden dividir en tres categorías. En la forma más leve los arcos vertebrales tuvieron una falla en su fusión, pero existe piel cubriendo el defecto y no hay tejido nervioso expuesto, forma que usualmente es asintomática.
La forma quística incluye los defectos cerrados con una obvia masa dorsal, como los mielomeningoceles cerrados y los meningoceles. En los meningoceles hay protrusión de las meninges, sin tejido nervioso. Cuando hay fibras nerviosas en el contenido del saco herniario, al defecto se le llama mielomeningocele. Las malformaciones espinales abiertas son las más comunes, siendo la forma clínica más severa, incluyen un defecto en la cobertura de piel y huesos, y la protrusión de un saco que contiene tejido nervioso. El déficit neurológico puede deberse tanto a la exposición de las raíces nerviosas como a la hidrocefalia asociada.
Estos defectos ocurren debido a la falla del cierre de los arcos vertebrales antes de las 6 semanas de gestación, secundario a una falla del desarrollo normal del tejido ectodérmico. Ocurren con mayor frecuencia en la región lumbosacra y se asocian a hidrocefalia debida a malformación de Arnold-Chiari tipo II en el 90% de los casos.
La incidencia varía según región geográfica, siendo de alrededor de 1 en 500 en el Reino Unido y 1 en 2.000 en Estados Unidos.
Anomalías cromosómicas, mutaciones genéticas, diabetes mellitus materna y la ingesta de teratógenos (como las drogas antiepilépticas), se asocian a alrededor del 10% de estos defectos, incluyendo la anencefalia y el encefalocele. Si uno de los padres o hermanos tiene uno de estos defectos hay un riesgo de 5-10% de recurrencia. El suplemento periconcepcional de la dieta materna con ácido fólico reduce a la mitad el riesgo de desarrollar estos defectos10,11.
El diagnóstico de espina bífida requiere del examen sistemático de cada vértebra, desde la región cervical a la sacra, tanto en el plano transversal como en el longitudinal. En el examen transversal, la vértebra normal tiene la apariencia de una circunferencia cerrada que va cubierta por piel, mientras que en la espina bífida la vértebra toma forma de U y se puede asociar a un saco quístico posterior a la columna. Si el saco no contiene estructuras neurales se llama meningocele y cuando incluye fibras nerviosas se llama mielomeningocele. Por último, existe una tercera forma de presentación llamada mielosquisis, en la que no existe un saco herniario, pero las fibras nerviosas están expuestas al líquido amniótico al no estar cubiertas de piel.
El diagnóstico se ve facilitado por el reconocimiento de anomalías asociadas tanto en la calota como en el cerebro. Estas anomalías incluyen el cabalgamiento de los huesos frontales (signo del limón), y la obliteración de la cisterna magna ya sea con la ausencia de cerebelo o la curvatura anterior anormal de los hemisferios cerebelares (signo de la banana). La ventriculomegalia en diferentes grados se asocia virtualmente a la totalidad de los casos al nacer, y a un 70% en el trimestre medio. Usualmente las alteraciones del cráneo y cerebro son más fáciles de visualizar que los defectos espinales.
El tamaño y nivel del defecto espinal determinan el pronóstico neonatal. Lesiones sobre L3 se asocian a paraplejia completa, las bajo L3 resultan en déficits motores y sensoriales variables en las extremidades inferiores. Las funciones intestinal y vesical se pueden también ver comprometidas.
Hernia diafragmática
La hernia diafragmática es una malformación congénita caracterizada por la protrusión hacia el tórax de contenido abdominal a través de un defecto del diafragma, este defecto es más frecuentemente posterolateral (hernia de Bochdalek) o retroesternal (hernia de Morgagni).
El desarrollo del diafragma usualmente se completa hacia la novena semana de gestación; si existe un defecto diafragmático ocurre la herniación de la vísceras abdominales hacia el tórax entre las 10 y 12 semanas de edad gestacional, cuando los intestinos retornan a la cavidad abdominal desde el cordón umbilical. Sin embargo, en algunos casos esto puede ocurrir tardíamente en el segundo trimestre.
La hernia diafragmática es usualmente un defecto esporádico; sin embargo, en aproximadamente el 50% de los casos se asocia a anomalías cromosómicas (especialmente trisomía 18, trisomía 13 y síndrome de Pallister-Killian), otras malformaciones o síndromes genéticos.
La incidencia es de aproximadamente 1 por cada 4.000 nacimientos, siendo 2 veces más frecuente en recién nacidos de sexo masculino.
El diagnóstico ecográfico de hernia difragmática se hace con la demostración de estómago o intestinos (90% de los casos) o hígado (50%) en el tórax acompañado de la lateralización hacia el lado contrario del contenido mediastínico. Se puede encontrar polihidroamnios (generalmente después de las 25-26 semanas de edad gestacional) en aproximadamente el 75% de los casos, lo que podría ser consecuencia de la dificultad de deglución del feto debido a la compresión esofágica por las vísceras herniadas.
Los principales factores pronósticos son el grado de hipoplasia pulmonar, hipertensión pulmonar secundaria y la coexistencia de otras anomalías. Aunque la hernia diafragmática aislada es un defecto anatómico fácil de reparar, se asocia a una mortalidad perinatal de alrededor del 50%, dada principalmente por hipoxemia secundaria a la hipertensión pulmonar resultante del desarrollo anormal del lecho vascular pulmonar.
Malformación pulmonar producto de una anormalidad del desarrollo de los bronquíolos terminales, dando origen a tumores benignos hamartomatosos o displásticos del pulmón.
Las malformaciones adenomatoídeas quísticas se desarrollan durante las primeras 6 semanas de gestación. Generalmente son unilaterales, afectando a sólo un lóbulo pulmonar, aunque también pueden ser bilaterales. Se clasifican en forma macroquística o CAM tipo I (quistes mayores a 5 mm de diámetro), mixta o CAM tipo II, o microquística o CAM tipo III (quistes menores a 5 mm de diámetro). Hay un 26% de asociación a otras malformaciones, siendo las más frecuentes el pectus excavatum, hidrops fetal y agenesia renal.
Tiene una incidencia de 1 en 400 nacimientos, encontrándose en igual proporción en recién nacidos de sexo masculino y femenino.
El diagnóstico ecográfico se hace a través de la visualización de tumores pulmonares hiperecogénicos en cualquiera de sus tres variantes, macroquística, mixta o microquística. La enfermedad microquística resulta en una hiperecogenicidad uniforme del parénquima pulmonar afectado. En la enfermedad macroquística, espacios quísticos únicos o múltiples se pueden visualizar en el tórax. Tanto la enfermedad microquística como la macroquística se pueden asociar con una desviación del mediastino; en la enfermedad bilateral, se puede ver el corazón comprimido, aunque no desviado. Cuando hay compresión cardíaca y de los grandes vasos torácicos, se desarrolla hidrops fetal. Otra característica común es el polihidroamnios que sería consecuencia de la dificultad del feto para tragar causada por la compresión esofágica, o por el aumento de producción de líquido amniótico por el tejido pulmonar anormal.
La enfermedad bilateral es letal, sea in utero, secundario al hidrops progresivo, o en el período neonatal. La enfermedad unilateral que no se asocia a hidrops es de buen pronóstico. En aproximadamente el 70% de los casos el tamaño del tumor se mantiene estable, en el 20% éste disminuye o desaparece, y en aproximadamente el 10% hay un aumento progresivo de la compresión mediastínica. En recién nacidos sintomáticos, se realiza toracotomía con lobectomía, con un 90% de sobrevida. El rol de la cirugía en los recién nacidos asintomáticos no está claro.
Malformaciones de la pared abdominal: onfalocele y gastrosquisis
El onfalocele se define como la malformación congénita caracterizada por la protrusión del contenido abdominal a través de la inserción umbilical y cubierta por una membrana que puede o no estar indemne.
La gastrosquisis es la malformación congénita en la cual existe una herniación visceral a través de un defecto en la pared abdominal lateral a un cordón umbilical intacto, y que no está cubierta por una membrana.
El onfalocele resulta de una falla en el proceso de regresión normal del intestino medio desde el conducto onfalomesentérico a la cavidad abdominal. El contenido abdominal, que puede incluir sólo intestino, o también hígado y bazo, se encuentran herniados a través del anillo umbilical, cubiertos por el peritoneo parietal y amnios. El onfalocele se asocia con otras malformaciones en más de la mitad de los casos, incluyendo malformaciones cardíacas, extrofia vesical, ano imperforado, defectos del tubo neural, labio leporino y hernias diafragmáticas. Además, aproximadamente el 25% de los casos se asocia a anomalías cromosómicas, especialmente las trisomías 13 y 18. En estos casos se debe descartar el Síndrome de Beckwith-Wiedemann, el que incluye la asociación de onfalocele con macrosomía, órganomegalia, macroglosia e hipoglicemia neonatal.
El onfalocele tiene una incidencia de 1 en 4.000 nacimientos, siendo 5 veces más frecuente en el sexo femenino.
El diagnóstico ecográfico del onfalocele se basa en la visualización del defecto anterior de la pared abdominal, el saco herniario con contenido abdominal y la inserción umbilical en la base del saco.
El pronóstico depende de la coexistencia de otras malformaciones o anomalías cromosómicas. Para lesiones aisladas la supervivencia tras corrección quirúrgica es del 90%.
En la gastrosquisis la rotación intestinal ocurrió normalmente, la evisceración intestinal ocurre a través de un defecto de la pared abdominal lateral, y usualmente a la derecha, de un cordón umbilical intacto. Las asas intestinales se encuentran descubiertas en el líquido amniótico, engrosando sus paredes y haciéndose edematosas.
La gastrosquisis tiene una incidencia de 1 en 4.000 nacimientos, encontrándose en igual proporción en fetos masculinos y femeninos.
Esta es una malformación esporádica, muy rara vez se asocia a anomalías cromosómicas, aunque otras malformaciones se encuentran presentes en 10-30% de los casos, especialmente atresias intestinales.
El diagnóstico ecográfico de gastrosquisis se basa en la demostración de un cordón umbilical normalmente situado y de asas intestinales herniadas flotando libremente en el líquido amniótico.
La sobrevida postoperatoria es aproximadamente del 90%, la mortalidad es usualmente consecuencia del síndrome del intestino corto, afección en la cual los niños requieren de nutrición parenteral total, y mueren, generalmente dentro de los primeros 4 años de vida, debido a insuficiencia hepática.
Atresia duodenal
La atresia duodenal consiste en la completa obliteración del lumen del duodeno, es la atresia congénita más común del intestino delgado.
A las 5 semanas de edad gestacional, el lumen duodenal se ve obliterado por epitelio proliferativo; la recanalización de éste se restablece para la semana 11; la falla de la vacuolización puede conducir a estenosis o atresia. También se puede ver obstrucción abdominal por compresión pancreática, en el páncreas anular, o por bandas peritoneales fibrosas.
La incidencia de atresia duodenal es de 1 en 10.000 nacimientos, y un tercio de ellos presenta trisomía 21.
El diagnóstico ecográfico de la atresia duodenal está dado por la visualización de la “doble burbuja” característica, dada por la dilatación gástrica y del duodeno proximal, lo que comúnmente se asocia a polihidroamnios. Esto es posible de diagnosticar desde las 20 semanas de edad gestacional, aunque generalmente se hace más evidente después de las 24 semanas.
La sobrevida en casos de atresia duodenal aislada es superior al 95% después de la reparación quirúrgica.
Riñón poliquístico
El término riñón poliquístico incluye 4 síndromes malformativos conocidos como Potter tipo I, II, III y IV.
El riñón poliquístico infantil o Potter tipo I, se caracteriza por riñones marcadamente aumentados de tamaño debido a múltiples quistes corticales y túbulos colectores dilatados. La enfermedad tiene un amplio espectro clínico dado por el daño renal y hepático, y se divide en tipos perinatal (el más común), neonatal, infantil y juvenil dependiendo del momento de inicio de la sintomatología y del grado de daño tubular.
Esta enfermedad se encuentra en 1 cada 30.000 nacimientos, en igual proporción entre los sexos femenino y masculino. Es una enfermedad autosómica recesiva. El gen responsable se encuentra en el brazo corto del cromosoma 6, por lo que el diagnóstico prenatal es posible durante el primer trimestre.
El diagnóstico ecográfico es posible en los tipos perinatal y posiblemente en el neonatal, y se basa en la visualización de riñones aumentados de tamaño bilateralmente, de parénquima uniformemente hiperecogénico. Frecuentemente se puede observar oligohidroamnios asociado, pero no siempre.
El tipo perinatal es invariablemente letal, sea in utero, o en el período neonatal debido a hipoplasia pulmonar. El tipo neonatal causa la muerte durante el primer año de vida debido a insuficiencia renal. Los tipos infantiles y juveniles cursan con insuficiencia renal crónica, fibrosis hepática e hipertensión portal.
La enfermedad renal multiquística o Potter tipo II, consiste en la displasia congénita de los riñones caracterizada por túbulos colectores con grandes dilataciones en forma no homogénea. Esto puede ocurrir uni o bilateralmente.
Aunque la patogenia es desconocida, se piensa que estaría causada por una falla temprana del blastema mesonéfrico o a una uropatía obstructiva; los túbulos colectores se hacen quísticos y el diámetro de los quistes va a determinar el tamaño renal, el que puede ser grande o pequeño.
Esta anomalía se encuentra en 1 de cada mil nacimientos, siendo más frecuente en los fetos de sexo masculino. Es la malformación quística renal más frecuente en el recién nacido.
En la ecografía los riñones se ven reemplazados por quistes de tamaño variable rodeados por estroma hiperecogénico. El desorden puede ser bilateral, unilateral o segmentario; si es bilateral se asocia a oligohidroamnios absoluto y ausencia de vejiga.
La enfermedad bilateral es invariablemente letal debido a la hipoplasia pulmonar asociada; la enfermedad unilateral, en cambio, es de buen pronóstico.
El síndrome de Potter tipo III corresponde al riñón poliquístico del adulto; es usualmente asintomático hasta la tercera o cuarta década de la vida, por lo que es improbable que se logre evidenciar la enfermedad durante la vida intrauterina. En esta enfermedad se pueden visualizar ambos riñones aumentados de tamaño en forma irregular, y en un tercio de los casos se acompaña de quistes hepáticos, pancreáticos, esplénicos o pulmonares. En un quinto de los casos, se pueden ver también aneurismas cerebrales. En la literatura hay pocos casos de diagnóstico ecográfico antenatal de esta enfermedad; sin embargo, se puede hacer el diagnóstico prenatal a través de la biopsia de vellosidades coriales.
El síndrome de Potter tipo IV o displasia quística secundaria a uropatía obstructiva es producido por una obstrucción en la vía urinaria fetal, produciendo una dilatación retrógrada progresiva, la que finalmente causa la displasia del riñón, con daño irreparable de éste. La mayoría de las veces la obstrucción es secundaria a una valva uretral posterior, aunque menos frecuentemente puede haber obstrucción a nivel de la unión ureteropélvica o uretero vesical.
El diagnóstico ecográfico se sospecha con la visualización de una dilatación de la pelvis renal en el corte transversal del abdomen (mayor a 10 mm). Esta obstrucción baja puede dar origen a megavejiga, megauréter, hidronefrosis y finalmente displasia renal (Potter tipo IV); dependiendo del momento y gravedad de la obstrucción ésta puede llevar a rotura del tracto urinario causando uroperitoneo secundario.
Displasias esqueléticas
Las displasias esqueléticas se encuentran aproximadamente en 1 cada 4.000 nacimientos; alrededor del 25% de los fetos afectados mueren en el período prenatal y aproximadamente el 30% lo hacen en el período neonatal.
De acuerdo con la momenclatura internacional para displasias esqueléticas, éstas se subdividen en 3 diferentes grupos:
Existe una gran cantidad de displasias esqueléticas, cada una con un riesgo de recurrencia específico, distinto fenotipo y distintas implicancias pronósticas. El conocimiento que existe de su expresión intrauterina es limitado. El descubrimiento incidental de una displasia esquelética en una ecografía de rutina necesita de un examen sistemático para llegar al diagnóstico correcto. Se deben evaluar todas las extremidades, tanto en su largo, forma y mineralización como en sus movimientos, y además, deben buscarse anomalías en otros órganos, especialmente en el cráneo, tórax y columna.
La malformación de alguna de las extremidades se denomina de acuerdo a la parte del miembro afectada. Micromelia es cuando existe acortamiento de toda la extremidad. Rizomelia es cuando el acortamiento afecta al segmento proximal de la extremidad. Finalmente, acromelia es cuando el acortamiento afecta al segmento distal de la enfermedad.
A grandes rasgos las displasias esqueléticas se pueden dividir en letales y no letales, como se muestra en la Tabla IV siguiente.
| Displasias | Tipos de Displasias | Incidencia |
| Letales
| Displasia tanatofórica Acondrogénesis Osteogénesis imperfecta tipo II Hipofosfatasia congénita Condrodisplasia punctata | 1 /10.000 1 /40.000 1 /60.000 1 /100.000 1 /110.000 |
| No letales | Acondroplasia heterozigótica Osteogénesis imperfecta tipo I Displasia toráxica asfíctica | 1/30.000 1/30.000 1/70.000 |
Tabla IV. Incidencia de las displasias esqueléticas.
Existen dos circunstancias en las cuales se debe buscar una displasia esquelética: en un paciente con el antecedente de tener un hijo afectado y que desea consejo genético y en caso de hallazgo de alguna anomalía en una extremidad durante un examen de rutina.
Es importante efectuar el diagnóstico antenatal dado que un buen número de las displasias esqueléticas son letales o se acompañan de un severo retardo mental, y otras se asocian a trombocitopenia y, por lo tanto, conocerla evitaría exponer al feto a un parto vaginal.
A continuación daremos una breve descripción de las displasias esqueléticas más frecuentes.
Displasia tanatofórica
Es la más frecuente de las displasias esqueléticas letales, con una frecuencia de 0,24 a 0,69 por 10.000 recién nacidos vivos (RNV), y parece heredarse en forma autonómica recesiva. Se caracteriza por rizomelia extrema, tronco de longitud normal, tórax angosto y cabeza grande con frente prominente. La tipo I se caracteriza por tener el fémur arqueado como auricular de teléfono y la tipo II por tener el cráneo en hoja de trébol y huesos rectos y cortos. El tórax pequeño determina la hipoplasia torácica, característica de la displasia tanatofórica. El diagnóstico suele hacerse en el tercer trimestre, aunque se han reportado casos más precoces. Esta condición es invariablemente letal.
Acondrógenesis
Es la segunda más frecuente de las displasias esqueléticas letales, su prevalencia es de 1 por 40.000 RNV y se transmitiría por nuevas mutaciones autosómicas dominantes. El diagnóstico antenatal se basa en el hallazgo de micromelia, falta de osificación vertebral y macrocefalia con grado variable de subosificación del calvarium. Los fetos pueden tener aspecto hidrópico dada la redundancia de partes blandas en relación a las extremidades cortas, y pobre osificación, pero no se observa acumulación de líquidos en las serosas.
Acondroplasia heterozigota
Es la más frecuente de las displasias esqueléticas no letales. Tiene una incidencia de 1 por 30.000 a 60.000 RNV, se puede transmitir en forma autonómica dominante (20% de los casos) o, en su mayoría, debido a una mutación espontánea (80% de los casos), siendo la edad paterna factor de riesgo para esta condición.
Se caracteriza por enanismo rizomélico, extremidades encorvadas y lordosis. Los huesos de manos y pies son pequeños (braquidactilia) y existe macrocefalia con aplanamiento del puente de la nariz, aplastamiento frontal y mandíbula ancha. El diagnóstico antenatal tiene la dificultad de que la alteración del crecimiento de los huesos largos no se expresa hasta el tercer trimestre.
La acondroplasia heterozigota es compatible con una vida normal, mientras que la variedad homozigota es letal.
A continuación se enumeran las malformaciones fetales que son invariablemente letales.
Anomalías congénitas letales
Anomalias congénitas con indicación de estudio genético8,9
Aquí se enumeran las anomalías congénitas que tendrían indicación de realización de estudio genético debido a su alta asociación con cromosomopatías.
Se ha establecido claramente la relación de la suplementación de la dieta con ácido fólico y la disminución tanto de la ocurrencia como de la recurrencia de malformaciones congénitas, especialmente en lo que se refiere a defectos del tubo neural. La recomendación actual para prevenir la ocurrencia de malformaciones es suplementar con 0,4 mg/día de ácido fólico desde 3 meses antes de la concepción hasta el término del período de organogénesis (12 semanas de edad gestacional); y, para prevenir la recurrencia, suplementar 4 a 5 mg/día por el mismo período de tiempo.
Las pacientes que se deben suplementar con mayor dosis son aquellas que tienen el antecedente de un hijo con alguna malformación mayor (en especial del SNC) y las que están recibiendo alguna terapia farmacológica que sea imposible de suspender y reconocidamente teratogénica (ej: anticonvulsivantes).
Los autores han completado el formulario de declaración de conflictos de intereses del ICMJE traducido al castellano por Medwave, y declaran no haber recibido financiamiento para la realización del artículo/investigación; no tener relaciones financieras con organizaciones que podrían tener intereses en el artículo publicado, en los últimos tres años; y no tener otras relaciones o actividades que podrían influir sobre el artículo publicado. Los formularios pueden ser solicitados contactando al autor responsable.
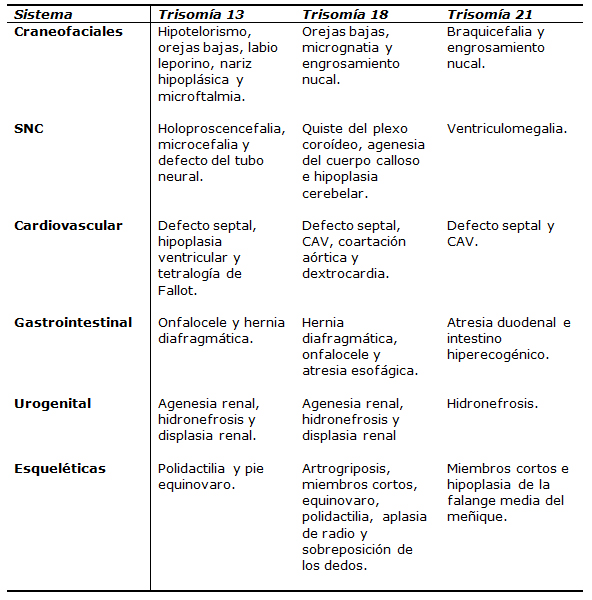 Tabla III. Malformaciones pesquisables por ultrasonido.
Tabla III. Malformaciones pesquisables por ultrasonido.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Congenital anomalies in conjunction with intrauterine growth restriction account for 50-60% of fetal mortality. This article describes major birth defects by systems; most can be diagnosed by ultrasound while others cannot. Some anomalies develop later in pregnancy and in some cases sonographic examination may suggest the presence of a nonexistent abnormality. Incidence, etiology and risk factors of congenital malformations are described, as well as the importance and characteristics of prenatal diagnosis and prevention through folic acid supplementation in the diet.
Autores:
Bernardita Donoso Bernales[1], Enrique Oyarzún Ebensperger[1]
Citación: Donoso B, Oyarzún E. congenital anomalies. Medwave 2012 Oct;12(9):e5537 doi: 10.5867/medwave.2012.09.5537
Fecha de envío: 25/9/2012
Fecha de aceptación: 26/9/2012
Fecha de publicación: 1/10/2012
Origen: solicitado
Tipo de revisión: sin revisión por pares
Citaciones asociadas
1. Impressum Oct;12(9) Medwave: cuerpo editorial de este número | Link |
2. Vázquez Martínez VR, Torres González CJ, Díaz Dueńas AL, Torres Vázquez G. Malformaciones congénitas en recién nacidos vivos. Medisur. 2014;12(1) | Link |
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Marden PM, Smith DW, McDonald MJ. Congenital anomalies in the newborn infant, including minor variations. A study of 4,412 babies by surface examination for anomalies and buccal smear for sex chromatin. J Pediatr. 1964 Mar;64:357-71 | CrossRef | PubMed |Chung CS, Myrianthopoulos NC. Congenital anomalies: mortality and morbidity, burden and classification. Am J Med Genet. 1987 Jul;27(3):505-23. | CrossRef | PubMed |Eydoux P, Choiset A, Le Porrier N, Thépot F, Szpiro-Tapia S, Alliet J, et al. Chromosomal prenatal diagnosis: study of 936 cases of intrauterine abnormalities after ultrasound assessment. Prenat Diagn. 1989 Apr;9(4):255-69. | CrossRef | PubMed |Burgoyne PS, Holland K, Stephens R. Incidence of numerical chromosome anomalies in human pregnancy estimation from induced and spontaneous abortion data. Hum Reprod. 1991 Apr;6(4):555-65. | PubMed |Leppig KA, Werler MM, Cann CI, Cook CA, Holmes LB. Predictive value of minor anomalies. I. Association with major malformations. J Pediatr. 1987 Apr;110(4):531-7. | CrossRef | PubMed |Pilu G, Nicolaides K, Ximenes R and Jeanty P. Diagnosis of fetal abnormalities, The 18-23 weeks scan. New York, USA: The Parthenon Publishing inc, 1999. American Institute of Ultrasound in Medicine, Standards AIUM Practice Guideline for the performance of the antepartum obstetrical ultrasound examinations. J Ultrasound Med. 2003 Oct;22(10):1116-25 | PubMed |American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 101: Ultrasonography in pregnancy. Obstet Gynecol. 2009 Feb;113(2 Pt 1):451-61. | PubMed |Berry RJ, Li Z, Erickson JD, Li S, Moore CA, Wang H, et al. Prevention of neural-tube defects with folic acid in China. China-U.S. Collaborative Project for Neural Tube Defect Prevention. N Engl J Med 1999; 341:1485. | CrossRef | PubMed |Hernández-Díaz S, Werler MM, Walker AM, Mitchell AA. Neural tube defects in relation to use of folic acid antagonists during pregnancy. Am J Epidemiol. 2001 May 15;153(10):961-8. | CrossRef | PubMed |
Parto prematuro






 Estudios originales
Estudios originales