Revista Biomédica Revisada Por Pares

Este texto completo es la transcripción editada y revisada de una conferencia dictada en el III Curso de Oncología Musculoesquelética y Sarcomas Óseos, Módulo I: Sarcoma de Ewing. Generalidades, organizado en Santiago por la Sociedad Chilena de Traumatología y Ortopedia del 16 al 18 de agosto de 2000.
Director Curso: Prof. Asistente Dr. Miguel Sepúlveda H.
Coordinadores: Dr. Orlando Wevar C., Dr. Julio Arriagada V.
El sarcoma de Ewing tiene una incidencia de 6 a 10% de los tumores óseos malignos primarios según lo descrito en la literatura, cifra que se repite en nuestro medio. Se define como un tumor compuesto de células pequeńas, sin producción de matriz, con grados variables de diferenciación neuroectodérmica.
La inmunohistoquímica nos revela la expresión de cementina, enolasa neuroespecífica, sinaptolisina, proteínas neurofilamentosas y otras esterasas.
Los estudios citogenéticos indican traslocación t(11;22) y (q24;q12) en 85% de los casos. La deleción del cromosoma 22 es menos frecuente, presentando además expresión de protooncogenes c-myc, n-myc, c-mit, c-mil/val-1 similares al PNET. (Son similares a los tumores neuroectodérmicos del hueso al tumor de Askin), y al osteosarcoma de células pequeńas y al neuroblastoma metastático.
Se presenta en la edad cercana a la adolescencia, en la que existe una gran cantidad de tumores de células pequeńas que generan problemas diagnósticos. Este es un tumor que no crea matriz y que puede tener una diferenciación neuroectodérmica variable, a veces casi inexistente, otras más evidente.
Hace unos 50 ańos Willis, un destacado patólogo australiano, observó que el sarcoma de Ewing presentaba unas rosetas y pensó que era una metástasis de neuroblastoma. No sólo Willis, sino que varios otros patólogos, plantearon la misma teoría.
Ahora sabemos que esto no es así, que presenta una diferenciación neuroectodérmica y que su parentesco es mucho más estrecho con el tumor neuroectodérmico periférico o PNET, con el cual comparte algo de morfología, si bien éste posee una mayor diferenciación neuroectodérmica. Los límites entre ambos no están claros en la literatura, la mayoría de los textos plantea que frente a un tumor con más de dos marcadores neuroectodérmicos, se debería pensar en un PNET más que en un Ewing. Además, el PNET presenta más rosetas en la histología. Por lo tanto, el tumor debería clasificarse nosológicamente como PNET mientras mayor diferenciación neuroectodérmica posea; no todos están de acuerdo con esta postura.
Debido a que el sarcoma de Ewing no presenta a la microscopía de luz ninguna diferenciación neuroectodérmica típica, salvo en ciertas ocasiones las rosetas, la clave está en la inmunohistoquímica, disponible en la mayoría de los laboratorios, al permitirnos una aproximación diagnóstica mucho más precisa.
La traslocación t (11;22) es propia del sarcoma de Ewing (85% de los tumores) y de otros tumores de tejidos blandos; también es propia de los PNET, del osteosarcoma de células pequeńas y del sarcoma de células claras de los tendones y membrana sinovial.
El sarcoma de Ewing expresa proteínas relacionadas con protooncogenes tales como c-myc, muy utilizado entre los patólogos ya que se traduce en una proteína de la superficie celular emparentada con factores de crecimiento de tipo insulina, que permite hacer una marcación bastante sensible y, por lo tanto, un diagnóstico más preciso del sarcoma de Ewing.
Otrora este diagnóstico se hacía por descarte, eliminando la posibilidad de tumores primarios desconocidos (hepatoblastomas, neuroblastomas, nefroblastomas, linfoma linfoblástico, etc.), todos los cuales poseen marcadores aunque no son específicos. A modo de ejemplo, el osteosarcoma no tiene marcadores propios y sólo presenta una proteína llamada cementina en su citoesqueleto, la cual ayuda al diagnóstico; sin embargo, el sarcoma de Ewing también presenta a veces la cementina y sólo pocas veces la enolasa neuroespecífica, de modo que éstos no eran marcadores totalmente confiables.
Actualmente, gracias a esta proteína y con la ayuda de varios marcadores existentes en el mercado, se puede realizar un mejor diagnóstico, ya que permite diferenciarlo, por ejemplo, del neuroblastoma metastásico, que posee proteínas similares. No obstante, como el comportamiento clínico y la terapia son similares en ambos tumores, no se ha diferenciado tajantemente uno de otro.
En la experiencia de Dalin podemos observar las localizaciones más frecuentes del sarcoma de Ewing en un universo de 300 casos: axial (costillas, sacro, pelvis) y tronco, ambas de mal pronóstico; la localización en las extremidades, tanto metafisiaria como diafisiaria, tiene mejor pronóstico; en las epífisis se ve rara vez. Vemos pocos casos en los pies, a diferencia de lo descrito en radiología y en la literatura.
Se puede apreciar una imagen radiológica típica ubicada en un hueso largo, donde se observa un compromiso extenso a nivel metafisiario y diafisiario, tanto de hueso como de partes blandas; son imágenes evidentemente muy agresivas.
Histología: formas típicas
La variedad más frecuente y clásica es la de células pequeńas, del tamańo de un linfocito. También existen de células grandes, de una vez y media el tamańo de un linfocito; ambas con la presencia característica de glucógeno y ausencia de estroma. En una revisión personal con microscopía de luz, el 51% tenía glucógeno, pero con microscopio electrónico esto sube a alrededor de 90%.
Formas atípicas
Se caracterizan por ausencia de glucógeno y estroma intercelular, pueden existir células fusadas y, en algunos casos, puede haber diferenciación rabdomioblástica, vascular y excepcionalmente, en roseta y ganglionar, con distintos patrones, entre ellos el lobulillar. El diagnóstico diferencial histológico se hace con osteosarcoma de células pequeńas, condrosarcoma mesenquimático, neuroblastoma metastásico, tumor de células pequeńas neuroectodérmicas de la región toracolumbar (tumor de Askin).
En los preparados histológicos se observa en los bordes una imagen de células pequeńas con fenómeno de invasión hacia los límites de la lesión, entre los espacios medulares preexistentes, sin deformar muy extensamente la morfología; sin embargo, puede existir un fenómeno de osteogénesis reactiva en respuesta a la destrucción causada por el tumor. En una imagen clásica vemos el citoplasma homogéneo sin estroma, salvo en los grandes vasos, el núcleo con límites tenues e imprecisos, sin citoplasma entre medio, la cromatina bastante fina de aspecto homogéneo, sin un nucléolo prominente, con zonas de meiosis muy variables; es una célula bastante frágil. A mayor aumento, los límites celulares se hacen prácticamente invisibles a la microscopía corriente, el núcleo se ve relativamente homogéneo, y en la cromatina, a pesar de ser maligno, no suelen verse figuras mitóticas.
Existen distintos patrones: difuso, aparentemente de mejor pronóstico; lobulillar, de mal pronóstico; y en filigrana, en seudo rosetas y endotelial, también de pronóstico adverso. Se trató de establecer una relación entre patrones y pronósticos, pero con el advenimiento de la quimoterapia esto quedó sin mayor significación.
Dificultades diagnósticas
Más allá del gran número de tumores de células pequeńas que presentan los nińos, hay dificultades inherentes a la calidad de la muestra. Estas dificultades se presentan con mayor frecuencia en los tumores de tejido óseo, que tienen menor accesibilidad por estar insertos dentro de un tejido firme, no así en un tumor extendido a tejidos blandos, donde la toma de muestra no presenta estas dificultades.
Se pueden seńalar las siguientes dificultades:
Disponemos de distintos tipos de tinciones para identificar distintos componentes de la célula. Por ejemplo, la tinción de retículo nos sirve para evidenciar el estroma, que en el sarcoma de Ewing es muy escaso, salvo rodeando a los vasos mayores. La tinción de PAS dando un color magenta, pone en evidencia el glucógeno del citoplasma de algunos tumores.
La microscopía electrónica pone en evidencia que tanto el PNET como el Ewing carecen de citoesqueleto, tienen pobreza de organelos, el 75% posee glucógeno, son muy uniformes en su contorno nuclear y celular, y no tienen estroma. Si uno cultiva las células se puede obtener diferenciación neuroectodérmica con gránulos de secreción neuroectodérmica, aunque no es lo habitual.
Diagnóstico diferencial de sarcoma de Ewing
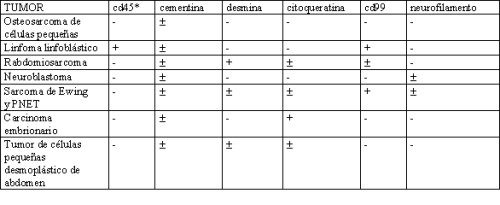
Aquí tenemos una lista no exhaustiva de tumores de células pequeńas y azules con los cuales se debe hacer el diagnóstico diferencial:
Inmunohistoquímica de tumores de células pequeńas, redondas y azules
+ : casi siempre positivo
± : sólo a veces positivo
- : casi siempre negativo
* Cd45 es el antígeno leucocitario común y Cd99 es la mutación de c-myc
Resumen de la inmunohistoquímica
En este resumen queda en evidencia el gran apoyo que significa la inmunohistoquímica para el diagnóstico:
CD99 (p30-32)
El gen myc2 está ubicado en la región seudoautosómica del brazo corto de los cromosomas humanos X e Y. Codifica para una proteína de superficie de 30 a 32 kd, y ha sido referido por varios autores como el antígeno de superficie celular del sarcoma de Ewing. La proteína se ha denominado myc2, p30-32 y, recientemente, se clasifica como CD99. Se ha desarrollado un set de anticuerpos monoclonales, incluyendo o13, hba71y 12e7(este último originalmente considerado como un antígeno característico de la leucemia), todos los cuales reconocen diferentes epitopos de CD99. Macroscópicamente observamos que la proteína de superficie da un halo café en la superficie celular.
Esta proteína de superficie está relacionada con antígenos de superficie de tipo insulínico; es un marcador altamente sensible para un subgrupo de tumores de células pequeńas redondas y azules de los nińos, particularmente sarcoma de Ewing, tumor neuroectodérmico periférico (PNET), linfoma linfoblástico y leucemia linfoblástica. Sin embargo, debe usarse como parte de un panel de anticuerpos para analizar estos tumores, ya que es compartida por varios de ellos y otros como el meningioma y el tumor fibroso solitario, que no entran en el diagnóstico diferencial del sarcoma de Ewing.
La proteína de membrana presenta un halo circular de color café. En este caso se ocupa un radioaislador alrededor de la célula para expresar la positividad para la proteína c-mic, cuyos valores para los distintos tumores son: linfoma linfoblástico 93%, rabdomiosarcoma 20%, sarcoma sinovial 50%, tumor insular 50%, tumor de células granulomatosas 50%, hemangiopericitoma 66%. Se encuentra casi siempre negativo en el neuroblastoma y en el carcinoma embrionario. Esto sirve para descartar a los del tercer grupo, mientras que los del primero con el segundo se deben descartar con la batería de la inmunohistoquímica.
La desmina se encuentra en un 15% de los Ewing-PNET. Es importante para el diagnóstico diferencial en los nińos, ya que está presente en más del 95% de los rabdomiosarcomas de cualquier tipo, igual que en el tumor de Wilms y en el tumor desmoplásico de células redondas, cuyo tratamiento quimioterápico es totalmente opuesto.
Alteraciones citogenéticas compartidas por Ewing y PNET
El sarcoma de Ewing comparte con el PNET alteraciones citogenéticas claves que se pueden estudiar en el laboratorio. Se observa una traslocación t(11;22)(q24;q12) más frecuente, presente en el 85% de los Ewing; en el otro 15%, se ve la traslocación alternativa t(21;22), o una reordenación (deleción) del cromosoma 22, del 22 q12, que da como resultado transcritos híbridos del gen ews con el gen fli-1 o erg, que pueden detectarse por estudios moleculares (RT-PCR). La posibilidad de encontrar tales transcritos híbridos está en el rango del 95%.
La relación entre esta traslocación característica, la formación de los transcritos híbridos y de la proteína, y la expresión de CD99, no está clara en este momento. La proteína depende de la mutación de c-myc, que está en los cromosomas X e Y en la región seudoautosómica; en cambio esta otra proteína está en el cromosoma 11-22, por lo que no está clara la relación entre esta traslocación y esta proteína; más bien está relacionada con un factor de crecimiento insulínico.
La traslocación alternativa ocurre en los cromosomas 21-22 en alrededor del 10% de los casos. El gen erg del cromosoma 21 se fusiona con el ews del 22, de carácter altamente oncogénico. La proteína erg que viene del cromosoma 21 pertenece a la familia ets de factores de transcripción, similar al fli-1 del cromosoma 11, de modo que la proteína de fusión producida por t (21-22) más frecuente, tiene un efecto tumorogénico similar a la t (11-22) y da lugar a un tumor histológicamente idéntico. Todo esto se puede detectar por inmunofluorescencia in situ o por PCR, que pueden detectar el tumor en pequeńas muestras de tejido: fli- ews, traslocación cromosómica 11- 22, o erg-ews, traslocación cromosómica 21-22.
Anatomía patológica: evaluación de la necrosis postquimioterapia
Otro aporte de la anatomía patológica es la evaluación del comportamiento del tumor frente a la quimioterapia. Si se dispone de una muestra podrá determinarse si está necrosada o no, si está fibrótica o no lo está; pero si se dispone de la pieza completa se puede determinar el porcentaje de necrosis y reemplazo por fibrosis o por tejido de cicatrización, y según eso se establece el porcentaje de respuesta a la terapia. Este es uno de los factores pronósticos más importantes en el manejo del sarcoma de Ewing.

 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el III Curso de Oncología Musculoesquelética y Sarcomas Óseos, Módulo I: Sarcoma de Ewing. Generalidades, organizado en Santiago por la Sociedad Chilena de Traumatología y Ortopedia del 16 al 18 de agosto de 2000.
Director Curso: Prof. Asistente Dr. Miguel Sepúlveda H.
Coordinadores: Dr. Orlando Wevar C., Dr. Julio Arriagada V.
Expositora:
Virginia Martínez[1]
Citación: Martínez V. Pathology of Ewing's sarcoma. Medwave 2001 Mar;1(03):e3476 doi: 10.5867/medwave.2001.03.3476
Fecha de publicación: 1/3/2001
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión







 Estudios originales
Estudios originales