Revista Biomédica Revisada Por Pares

Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso Actualización en Farmacología, organizado por Medwave Capacitación Limitada y la Unidad de Capacitación del Hospital Padre Hurtado entre los días 9 y 30 de junio de 2003. Director del Curso: Dr. Juan Diego Maya.
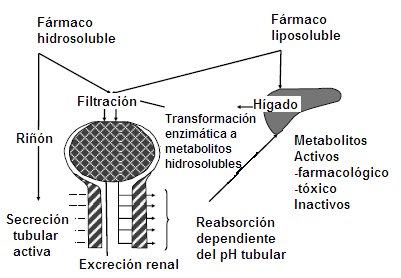
Los procesos de eliminación de un fármaco incluyen dos situaciones fisiológicas: la biotransformación y la excreción. La biotransformación ocurre preferentemente en el hígado, pero no exclusivamente, ya que el intestino, la placenta y el pulmón pueden participar de dicho proceso, el que tiene como objetivo la transformación enzimática de cualquier sustancia exógena al organismo en metabolitos hidrosolubles para facilitar la excreción renal, ya que mientras más liposoluble es un fármaco, más tiempo permanecerá en el organismo. Por ejemplo, el insecticida organofosforado DDT es tan liposoluble que permanece en el hígado sin ser metabolizado, por lo que no se elimina.
Un fármaco hidrosoluble se puede filtrar o secretar a nivel del túbulo renal y al llegar a la orina no se reabsorbe, por lo que se elimina. La transformación enzimática en metabolitos hidrosolubles puede originar metabolitos farmacológicamente activos; por ejemplo, el diazepam tiene una vida media de 36 horas, pero además tiene un metabolito activo cuya vida media es de 100 horas, por lo que también se debe metabolizar dicho metabolito a nivel hepático para que cese el efecto del fármaco. Cuando un paciente ingiere una sobredosis aguda de paracetamol, éste destruye las reservas de glutatión hepático, lo que genera la producción de metabolitos toxicológicamente activos que provocan una necrosis hepática aguda. El resto de los fármacos y sus metabolitos son inactivados a nivel hepático (Fig. 1).
Figura 1. Eliminación de fármacos. Remoción irreversible del fármaco del cuerpo por todas las rutas.
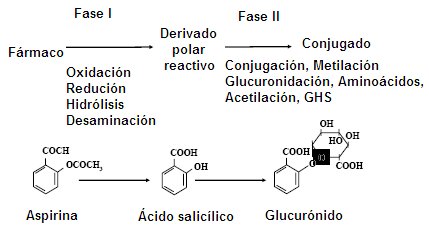
El metabolismo es un proceso secuencial que ocurre en dos fases, donde intervienen dos grupos enzimáticos. Durante el proceso enzimático de fase I los fármacos se convierten en sustancias muy reactivas; aquí el citocromo P-450 hepático juega un rol fundamental. El derivado polar reactivo de la fase I será el sustrato de las enzimas de fase II, en la cual puede experimentar procesos de glucoronidación, acetilación y metilación, además de adición de aminoácidos o glutatión. Cuando el fármaco reactivo se conjuga con una de estas moléculas pierde reactividad y liposolubilidad, es decir, se convierte en una sustancia hidrosoluble que será fácilmente eliminada. Existen fármacos que no se metabolizan, como es el caso de la penicilina, que se excreta del organismo tal cual entra. Los antiinflamatorios no esteroidales (AINES) se excretan en 98% sin alteración y los salicilatos pueden sufrir algún grado de glucoronidación (Fig. 2)
Figura 2. Fases del metabolismo de los fármacos.
El citocromo P-450 es un conjunto de proteínas con actividad enzimática oxidante que se ubican en el retículo endoplasmático liso (microsomas) del hepatocito. Este sistema es inespecífico y fácilmente inducible, es decir, su actividad aumenta en presencia de una sustancia; es por esto que un paciente fumador requiere cantidades mayores de aminofilina que un paciente no fumador, debido a que sus enzimas hepáticas están inducidas. A su vez, el citocromo P-450 es potencialmente saturable, porque existe una cantidad finita de enzima; esto significa que si se aporta un exceso de sustancia, el sistema se satura. También es fácilmente inhibible; por ejemplo, la eritromicina es capaz de inhibir las enzimas hepáticas que metabolizan el cisapride, fármaco que por sí solo tiene toxicidad cardíaca, motivo por el cual la combinación cisapride-eritromicina no se debe usar, sobre todo en nińos, porque puede producir arritmias fatales.
Los factores que determinan la eficiencia del hígado para eliminar fármacos son: la cantidad de fármaco que llega al hígado en la unidad de tiempo, lo que depende del flujo sanguíneo y de la concentración del fármaco en la sangre; la concentración del fármaco libre, es decir, el que no está unido a proteínas plasmáticas; y la actividad de los sistemas enzimáticos implicados en la biotransformación.
Figura 3. Cálculo del clearance de un fármaco.
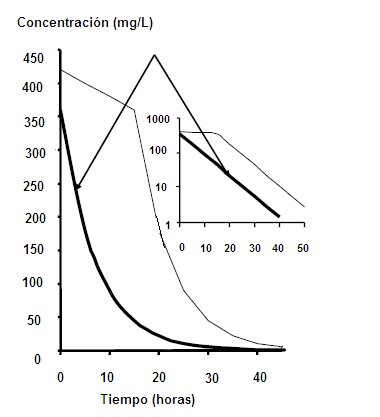
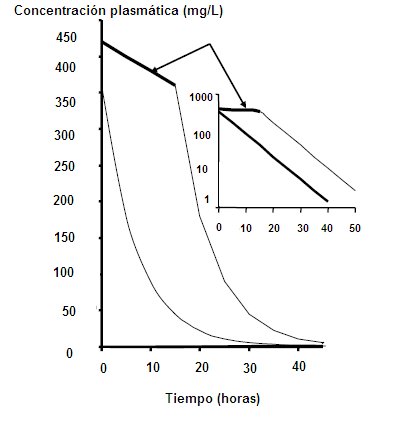
En condiciones terapéuticas, y considerando que un órgano es capaz de manejar un volumen fijo de plasma, el porcentaje de fármaco que será eliminado en función del tiempo es siempre constante. Por ejemplo, si existe en el plasma una cantidad x/ml de medicamento, en la siguiente unidad de tiempo habrá 50% menos, es decir, 0,5 x/ml y en la siguiente unidad de tiempo habrá 25%, o 0,25 x/ml, es decir, una disminución de 50% de lo anterior. Por lo tanto, la velocidad de desaparición del fármaco en el organismo es rápida, es decir, con cinética de orden 1 (Fig. 4).
Figura 4. Cinética de orden 1
Cuando el sistema de eliminación está saturado, es decir, cuando existe exceso de fármaco y el sistema tiene que manejar una cantidad fija de fármaco cada vez, en este caso la caída de la concentración plasmática del fármaco es constante, una pequeńa cantidad por vez. Si tenemos una concentración plasmática de 500 mg/ml y el sistema metabólico de excreción se encuentra saturado, sólo será capaz de manejar una cantidad constante, por ejemplo 15 mg/hora. A la hora habrá 485 mg/ml; entonces, en 2 horas sólo habrán salido 30 mg, cuando en otras condiciones habrían salido 400. Esto corresponde a una cinética de orden 0, donde el sistema está colapsado. Este tipo de cinética se ve cuando un paciente está intoxicado con Aspirina o con etanol; por este motivo la alcoholemia se extrapola al momento del accidente, ya que se pierde una cantidad pequeńa, con una relación lineal (Fig. 5).
Figura 5. Cinética de orden 0.
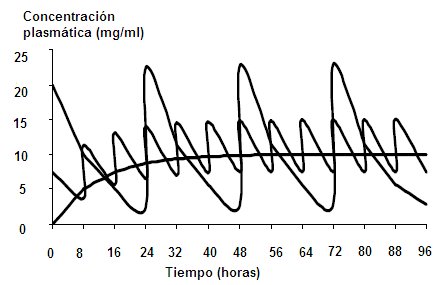
Figura 6. Fluctuación de las concentraciones plasmáticas respecto del intervalo y dosis de administración.
Los objetivos de los regímenes de dosificación son: mantener concentraciones terapéuticas de un fármaco en el estado estacionario; individualización de la terapia; mantener la concentración plasmática dentro de rangos apropiados; se utiliza en forma preferente para fármacos con margen terapéutico estrecho como los anticonvulsivantes, antiarrítmicos, digoxina y teofilina. La teofilina y la carbamazepina son capaces de inducir su propio metabolismo mediante la inducción enzimática del citocromo P450, por lo cual se debe establecer un régimen de dosificación considerando este hecho y aumentar las dosis del fármaco cuando se requiera.
La dosis de mantención es la cantidad de fármaco más apropiada para su administración, en infusión continua o en forma intermitente, con el objetivo de mantener el estado estacionario. El valor calculado depende del clearence del fármaco; cuando la administración es vía oral, la dosis dependerá de la vida media del fármaco y se obtiene del producto del clearence por la concentración plasmática. Si se multiplica la dosis de mantención por la vida media, el resultado es el intervalo de administración del fármaco.
La dosis de carga es la cantidad de fármaco necesaria para alcanzar en forma rápida la concentración deseada, dentro del rango terapéutico. Significa que se llenarán los depósitos físicos con el fármaco en forma aguda. Siempre existe el riesgo de alcanzar concentraciones plasmáticas tóxicas de una sola vez, por esta razón la digoxina se administra fraccionada y no en bolo. La dosis de carga depende del volumen de distribución del fármaco.


 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso Actualización en Farmacología, organizado por Medwave Capacitación Limitada y la Unidad de Capacitación del Hospital Padre Hurtado entre los días 9 y 30 de junio de 2003. Director del Curso: Dr. Juan Diego Maya.
Expositor:
Juan Diego Maya[1]
Citación: Maya JD. Pharmacokinetics: elimination. Medwave 2007 Jun;7(5):e3450 doi: 10.5867/medwave.2007.05.3450
Fecha de publicación: 1/6/2007
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión







 Estudios originales
Estudios originales