Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso de Genética, organizado por el Instituto de Ciencias Biomédicas de la Universidad de Chile entre los meses de marzo y julio de 2004.
Edición Científica: Prof. Laura Walker Bozzo, Profesora Encargada de Curso; Prof. Luisa Herrera Cisterna, Coordinadora de Curso.
El Cáncer Colorrectal Hereditario No Polipósico (HNPCC), también conocido como Síndrome de Lynch, es la forma más común de cáncer colorrectal hereditario; su incidencia corresponde a un 10% a 1% de los cánceres de colon. La aparición de esta enfermedad se produce durante la tercera y cuarta década de la vida, aunque no es extrańo encontrar individuos que lo presenten en edades más tempranas o más tardías.
En la actualidad se acepta la existencia de tres vías distintas que conducen a la aparición de cáncer colorrectal (CCR). Dos de ellas tienen un componente familiar hereditario plenamente establecido; son el HNPCC o síndrome de Lynch y la poliposis adenomatosa familiar (PAF). La tercera vía no se ha podido relacionar con mecanismos de transmisión hereditaria y sería el CCR esporádico.
El HNPCC se diferencia de los otros tipos de cánceres de colón porque los pacientes presentan muy pocos o ningún pólipo. Los tumores en pacientes con HNPCC comienzan presentándose como adenomas o tumores benignos, los que pueden transformarse en carcinomas o tumores malignos. Los individuos que presentan HNPCC están más predispuestos a desarrollar cáncer a nivel de endometrio, ovario, estómago, intestino delgado, tracto hepato-biliar, tracto urinario, cerebro y piel.
En este trabajo se explicarán las bases genéticas que determinan esta enfermedad, sus características clínicas, su patogénesis y su incidencia y manejo.
Muchos pacientes con cáncer colorrectal no presentan síntomas en estadios tempranos, sino hasta que la enfermedad está bastante avanzada. Los siguientes síntomas son los más frecuentes:
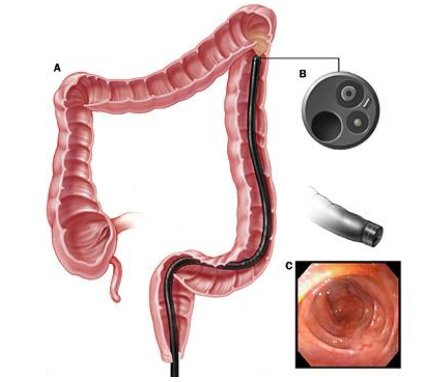
En la figura 1 se muestra el sigmoidoscopio flexible, que es un tubo flexible y corto que contiene iluminación y transmite imágenes del interior del colon y el recto hacia un monitor. Con este instrumento se puede efectuar una colonoscopía de todo el intestino grueso, en la que se debería encontrar muy pocos o ningún pólipo, característica que lo diferencia de otros tipos de cánceres de colon.
Figura 1. Colonoscopía con sigmoidoscopio flexible. A: Posición del sigmoidoscopio flexible dentro del colon; B: Extremo terminal del sigmoidoscopio; C: Imagen de endoscopía (1).
Algunos pacientes con el síndrome de HPNCC padecen también de cáncer de otros órganos, principalmente endometrio, estómago y rińón. En el caso de padecer cáncer de endometrio la paciente presentará dolores pélvicos o sangrado vaginal fuera del periodo menstrual; si tuviese cáncer de estómago, el paciente presentará alteraciones digestivas, dolor abdominal, vómitos con sangre, acidez, heces oscuras y fétidas y decaimiento; finalmente, si presentase un cáncer en el rińón, este se manifestaría por la presencia de sangre en la orina y dolores de espalda. La presencia de estos síntomas no implica que el paciente padezca de HNPCC, pero se hace recomendable que se realice un control médico (1).
El HNPCC es una enfermedad hereditaria que presenta un patrón de herencia autosómico dominante, es decir, si se presenta en un individuo uno de los alelos del gen anormal heredado de uno de los padres, éste es capaz de causar la enfermedad (2, 3).
El HNPCC generalmente tarda varios ańos en aparecer en la vida del individuo. La edad promedio de manifestación de la enfermedad es de 44 ańos, aunque como se mencionó anteriormente, no es raro encontrar individuos que expresan la enfermedad a edades más tempranas, inclusive alrededor de los 20 ańos (4).
El HNPCC es producto preferentemente de mutaciones en los genes que codifican para proteínas que participan en la reparación de bases mal apareadas ( mismatch repair o MMR). El proceso de MMR es precozmente activado por p53, una proteína que cumple una amplia gama de funciones, en especial, en el control del ciclo celular y reparación de mutaciones. Además participan en este proceso otras nueve proteínas codificadas por genes localizados en distintos loci: MSH1, MSH2 (2p21-p22), MSH3, MSH6 (2p16), MLH1 (3p21.3), MLH2, MTH, PMS1 (2q31.1) y PMS2 (7p22) (5).
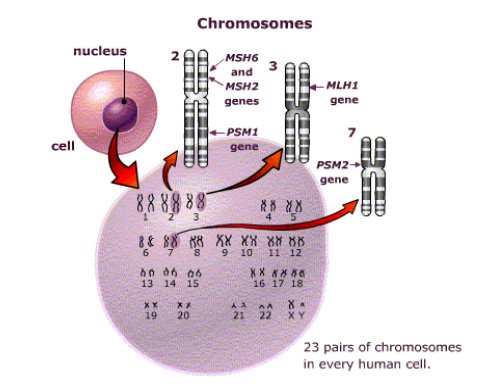
El HNPCC está asociado con mutaciones en uno o más de cuatro de estos genes: MLH1, MSH2, MSH6 y PMS2, siendo las mutaciones en MLH1 y MSH2 las más frecuentes ya que están presentes en cerca de 90% de los casos de HNPCC. Estos dos genes representan la base sobre la cual se realizan los estudios genético clínicos para determinar HNPCC en un individuo (6, 7). En los últimos ańos se ha descrito un aumento de casos de HNPCC debido a mutaciones en MSH6 (aproximadamente 7%-10% de las familias con HNPCC). Es por esto que podemos afirmar que el HNPCC presenta heterogeneidad genética, ya que puede ocurrir por existir mutaciones en genes no alelos, localizados en distintos loci, y/o porque existen diferentes tipos de mutaciones (inserción, deleción, traslocación, etc.) en genes alelos. En la figura 2 se ilustran estos conceptos.
Figura 2. Localización cromosómica de los genes asociados a MMR y a la aparición
de HNPCC.
Esta enfermedad presenta una penetrancia incompleta (2), es decir, dentro del total de las personas que presentan el genotipo que codifica para HNPCC, no todas presentan la enfermedad, de modo que existen individuos que, pese a presentar el genotipo “mutado” que normalmente desencadenaría la enfermedad, no la expresan fenotípicamente. Así, la mayoría de los individuos con HNPCC lo han heredado de sus padres, pero en muchos casos el fenotipo no se expresa en éstos, debido a la penetrancia incompleta del HNPCC y también a la variabilidad de la edad en que la enfermedad se desarrolla. En este caso, entonces, la dominancia del gen está supeditada a la penetrancia incompleta del mismo. Así, un individuo, pese a presentar el genotipo dominante, que lo hace más susceptible a desencadenar la enfermedad, puede presentarla en grado leve o simplemente no presentarla y ser un “portador” de ella.
El fenómeno de penetrancia incompleta se puede explicar suponiendo que el individuo “portador”, si bien tiene mutados ambos alelos de uno de los genes críticos para HNPCC, estos están hipermetilados, lo que los inactiva genéticamente, impidiendo su transcripción. Si los demás genes MMR están intactos (no mutados), el individuo contaría con casi toda la capacidad reparadora de los genes MMR. En esta situación, el individuo portador en cuestión puede no presentar HNPCC o puede presentar una forma leve de la enfermedad.
Como se describió previamente, los genes asociados al Síndrome de Lynch pertenecen a una gran familia de genes encargados de la reparación de los errores de apareamiento del DNA. La consecuencia de la inactivación de estos sistemas de reparación es la falta de reparación de los errores en el apareamiento de bases, produciéndose una acumulación de mutaciones a lo largo del tiempo que, a su vez, es la causante de la inestabilidad genómica presente en este tipo de patologías y de la aparición de células tumorales.
Como se mencionó previamente, la edad promedio de manifestación de la enfermedad es de 44 ańos. Esto se debe, en parte, a que se requiere de un tiempo considerable para que se acumulen mutaciones suficientes para desencadenar la enfermedad. La mayoría de las mutaciones que generan HNPCC se producen en el gen MLH1 (50%), lo siguen en frecuencia los genes MSH2 con 40% y MSH6 con 10% a 7%, y la frecuencia más baja de mutaciones se da en el gen PMS2, que se presenta mutado en menos de 2% de los casos de HNPCC (8).
Las proteínas codificadas por el gen MSH2 forman heterodímeros con los productos de MSH6 y MSH3. Este complejo molecular se encarga de identificar los errores en el apareamiento durante la replicación mediante deslizamiento sobre las hebras de DNA (8). Por otra parte, el producto génico de MLH1 forma un heterodímero con el de PMS2 y esto coordina la unión de proteínas relacionadas directamente con los procesos de reparación, como endonucleasas, helicasas, antígenos nucleares de células en proliferación (PCNA), proteínas SSB (single-stranded binding protein) y DNA polimerasas, entre otras.
Un error en la formación del heterodímero encargado de la búsqueda de error en el apareamiento, o un error en la unión de este complejo a proteínas esenciales para la reparación del DNA, determinan fallas en la reparación. El heterodímero podría no encontrar los sitios con error o confundir el DNA normal con uno anormal, y en el caso que los encontrara, existe la posibilidad que su reparación se frustre porque las proteínas generadas por MLH1 y PMS2 no coordinen adecuadamente la secuencia de acción de las helicasas, endonucleasas, etc.
La comprensión de las funciones de las proteínas codificadas por los genes MLH1, MSH2, MSH6 y PMS2 permite entender como actúan las mutaciones de estos genes en el incremento del riesgo de contraer cáncer. Las proteínas codificadas por MLH1 y MSH2 juegan un rol importante en la detección de mutaciones, mientras que MSH6 y PMS2 tienen un papel directo en la reparación. Realizando estas funciones estas proteínas logran entonces suprimir la posibilidad de desarrollar un cáncer como resultado de acumulaciones de mutaciones en genes específicos en una célula determinada (8).
El diagnóstico de HNPCC puede realizarse a partir de dos procedimientos. Uno de ellos corresponde al criterio clínico de Amsterdam y el otro a un análisis genético-molecular; este último se utiliza para identificar mutaciones en alguno de los genes MMR. El criterio clínico de Amsterdam está vigente desde 1990 gracias al International Collaborative Group on HNPCC, con el propósito de identificar familias para estudios de investigación.
El criterio clínico de Amsterdam I especifica que para diagnosticar el HNPCC deben existir tres o más familiares con un diagnóstico confirmado de cáncer de colon y al menos uno de ellos debe tener relaciones de parentesco de primer grado con los otros dos. También tienen que estar afectadas dos generaciones consecutivas, uno o más de los cáncer de colon deben ser diagnosticados antes de los 50 ańos y los pólipos adenomatosos familiares (FAP) tienen que ser excluidos.
El criterio clínico de Amsterdam II establece que para diagnosticar el HNPCC deben existir tres o más familiares con un diagnóstico confirmado de un cáncer asociado a HNPCC (colorrectal, endometrial, intestinal, hepatobiliar, renal o ureteral) y al menos uno de ellos debe tener relaciones de parentesco de primer grado con los otros dos. También tienen que estar afectadas dos generaciones consecutivas, uno o más de los cáncer asociados a HNPCC deben ser diagnosticados antes de los 50 ańos y los pólipos adenomatosos familiares (FAP) tienen que ser excluidos.
Con el criterio clínico de Amsterdam I se verifica en un 61% la presencia de mutaciones en el gen MSH2 y en un 67% en el gen MLH1. Con el criterio clínico de Amsterdam II, la sensibilidad del diagnóstico aumenta a un 78%.
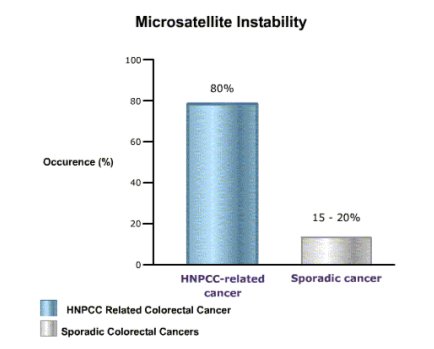
Existen diferentes metodologías genético-moleculares para diagnosticar el HNPCC. Una de ellas es la medición de la inestabilidad de los DNA microsatélites. Los DNA microsatélites, son un tipo de DNA no codificante, formado por secuencias repetitivas de nucleótidos. Estas secuencias repetitivas están usualmente expuestas a adquirir errores durante la replicación del DNA, errores que en personas con fallas en el MMR no pueden ser reparados, tal como se puede ver en la figura 3. Mientras más inestables sean los microsatélites más errores presentarán. Por esta razón, se utilizan marcadores para evaluar el grado de inestabilidad de los microsatélites (8).
Figura 3. Porcentaje de Inestabilidad de Microsatélites (MSI) en individuos con HNPCC y en individuos que desarrollan cáncer de forma esporádica (Fuente: American Medical Association, Chicago, Ill).
Existen dos métodos genético-moleculares que analizan los genes propiamente tales. Estos son: el escaneo de mutaciones y análisis de secuencias, y el análisis de deleciones y reordenamientos. En el primero de estos métodos se escanean los genes MLH1, MSH2 y MSH6 en busca de mutaciones, analizando sus secuencias nucleotídicas. La limitación de esta técnica es que no es capaz de detectar deleciones ni cambios en la localización de los genes. En el segundo procedimiento se utiliza la técnica de Southern blot, ya que esta permite identificar deleciones y reordenamientos de genes en el DNA.
Existe una estrategia, conocida como la pauta de Bethesda, para verificar la existencia de HNPCC en un individuo sospechoso de tener la enfermedad. Esta pauta establece que debe realizarse un análisis de inestabilidad de microsatélites siempre que se haya cumplido con uno o más de los criterios clínicos que propone. Estos criterios clínicos están muy relacionados con los criterios de Amsterdam.
Otra metodología genético-molecular a emplear para diagnosticar HNPCC es la inmunohistoquímica, en la que se controlan los productos de los genes MMR, los que están muy disminuidos en pacientes con HNPCC.
La mayoría de las estrategias de diagnóstico combinan los criterios clínicos (Amsterdam), con los genético-moleculares, existiendo de esta manera diversas combinaciones de técnicas para diagnosticar el HNPCC.
Existe también un test de diagnóstico prenatal para la detección de HNPCC, que consiste en el análisis de DNA a partir de células fetales obtenidas por amniocentesis entre las 16 a 18 semanas de gestación o por una prueba de vello coriónico entre las 10 a 12 semanas de gestación. Se requiere haber realizado la identificación de las mutaciones en los padres antes de efectuar el test de diagnóstico prenatal.
Existen restricciones para la detección de portadores de HNPCC en adultos jóvenes, que han sido establecidas por The American College of Medical Genetics y por The American Society of Human Genetics. Las pautas restrictivas de estas instituciones establecen que los individuos menores de 18 ańos no deben someterse a exámenes porque aún no tienen la mayoría de edad y no son capaces de tomar decisiones en forma independiente.
Antes de realizar pruebas de diagnóstico en adultos, se recomienda realizar orientación genética. Este proceso incluye la discusión de las implicancias clínicas y psicosociales de los resultados de las pruebas genéticas para el individuo y los miembros de la familia. Esto se debe hacer ya que los individuos a los cuales se les diagnostica HNPCC no tienen una buena calidad de vida posterior al diagnóstico y a veces esta mala calidad de vida se ve intensificada por los bajos niveles de apoyo social. Por estas razones se debe orientar a los padres con intenciones de tener hijos, ya que la probabilidad de que uno de sus hijos adquiera el HNPCC es del 50% y además es necesario advertir de las ventajas y desventajas de la realización de test genéticos (8).
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso de Genética, organizado por el Instituto de Ciencias Biomédicas de la Universidad de Chile entre los meses de marzo y julio de 2004.
Edición Científica: Prof. Laura Walker Bozzo, Profesora Encargada de Curso; Prof. Luisa Herrera Cisterna, Coordinadora de Curso.
Expositores:
Diego García Prado[1], Sebastián García Tobar[1], Aníbal García-Huidobro Cabrera[1]
Citación: García D, García S, García-Huidobro A. Hereditary nonpolyposis colon cancer: genetic aspects. Medwave 2005 Ene;5(1):e3371 doi: 10.5867/medwave.2005.01.3371
Fecha de publicación: 1/1/2005
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
de Leon MP, Pedroni M, Benatti P, Percesepe A, Di Gregorio C, Foroni M, et al. Hereditary colorectal cancer in the general population: from cancer registration to molecular diagnosis. Gut. 1999 Jul;45(1):32-8. | CrossRef | PubMed | PMC |Rodriguez-Bigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrom: Meeting Highlights and Bethesda Guidelines. J Natl Cancer Inst. 1997 Dec 3;89(23):1758-62. | CrossRef | PubMed |Aarnio M, Mecklin JP, Aaltonen LA, Nystrom-Lahti M, Järvinen HJ. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer. 1995 Dec 20;64(6):430-3. | CrossRef | PubMed |Peltomaki P, Vasen HF. Mutations predisposing to hereditary nonpolyposis colorectal cancer: Database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology. 1997 Oct;113(4):1146-58. | CrossRef | PubMed |Kowalski LD, Mutch DG, Herzog TJ, Rader JS, Goodfellow PJ. Mutational analysis of MLH1 and MSH2 in 25 prospectively-acquired RER+ endometrial cancers. Genes Chromosomes Cancer. 1997 Mar;18(3):219-27. | CrossRef | PubMed |Lin KM, Shashidharan M, Thorson AG, Ternent CA, Blatchford GJ, Christensen MA, et al. Cumulative incidence of colorectal and extracolonic cancers in MLH1 and MSH2 mutations carriers of hereditary nonpolyposis colorectal cancer. J Gastrointest Surg. 1998 Jan-Feb;2(1):67-71. | CrossRef | PubMed |Gruber SB, Kohlmann W. Hereditary Non-Polyposis Colon Cancer. J Natl Compr Canc Netw. 2003 Jan;1(1):137-44. | PubMed |






 Estudios originales
Estudios originales