Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso de Genética, organizado por el Instituto de Ciencias Biomédicas de la Universidad de Chile entre los meses de marzo y julio de 2004.
Edición Científica: Prof. Laura Walker Bozzo, Profesora Encargada de Curso; Prof. Luisa Herrera Cisterna, Coordinadora de Curso.
Existen dos tipos de distrofia miotónica (DM): tipo 1, que corresponde al 98% de los casos y que es causada por una expansión de la repetición del triplete CTG en el cromosoma 19, y tipo 2, que afecta al 2% restante, es conocida como PROMM (“proximal myotonic myopathia”) y su causa es la expansión de la repetición de los nucleótidos CCTG en el cromosoma 3. En este trabajo nos referiremos a la DM tipo 1, ya que es la más común.
La DM tipo 1 se caracteriza por una atrofia progresiva de los músculos, que pueden contraerse, pero encuentran dificultad para relajarse (miotonía). Los síntomas varían ampliamente y afectan a muchos sistemas, además del muscular.
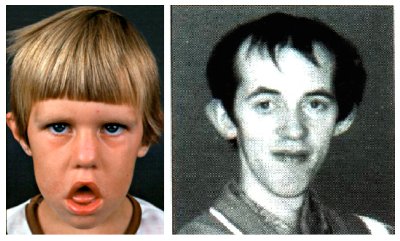
Los individuos afectados tienen un aspecto característico de “cara de cuchillo”, debido a la atrofia y debilidad de los músculos faciales (Figura 1). Los músculos del cuello están afectados precozmente, al igual que los músculos distales de los miembros, lo que puede dar lugar al “pie en péndulo”.
Figura 1. Fenotipo de distrofia miotónica. La debilidad de los músculos faciales confiere una expresión característica (facies), parecida a una máscara. Las mejillas se encuentran hundidas, la sonrisa invertida y se presenta ptosis palpebral (caída del párpado).
Los músculos proximales permanecen fuertes a lo largo de la enfermedad, aunque en muchos pacientes se produce atrofia y debilidad, preferentemente de los cuádriceps. El compromiso de la musculatura palatina, faríngea y lingual produce disartria, voz nasal y problemas de deglución. Algunos pacientes presentan debilidad en el diafragma y en los músculos intercostales, lo que causa insuficiencia respiratoria. La enfermedad se caracteriza también por debilidad facial y bulbar intensa e insuficiencia respiratoria neonatal.
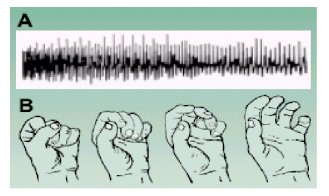
La miotonía puede demostrarse percutiendo la eminencia tenar, la lengua y los músculos extensores de la muńeca, lo que produce una relajación lenta de la mano tras cerrarla al máximo voluntariamente (Figura 2). El deterioro muscular avanzado dificulta más la detección de la miotonía.
Figura 2. (A) Miotonía en electromiografía (EMG). Se evidencia la lenta relajación del músculo después de una contracción máxima. (B) La miotonía se manifiesta como dificultad al abrir la mano (relajación) luego de una contracción voluntaria.
La mayoría de los pacientes con distrofia miotónica padece trastornos cardiovasculares, como bloqueo cardíaco de primer grado, bloqueo cardíaco completo o un compromiso más extenso del sistema de conducción. También es frecuente el prolapso de la válvula mitral.
Otras características asociadas a la DM tipo 1 son déficit intelectual, hipersomnia, cataratas subcapsulares posteriores, atrofia gonadal, resistencia a la insulina y reducción de la motilidad esofágica y cólica.
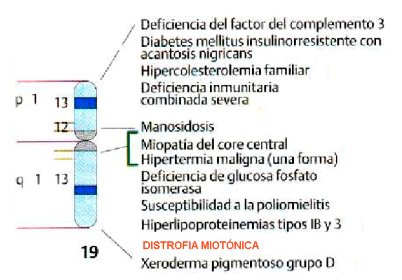
La DM tipo 1 es una enfermedad heredable en forma autosómica dominante. La anomalía se localiza en el brazo largo del cromosoma 19, banda 13.3 (19q13.3), que se muestra en la Figura 3, donde se encuentra el gen codificante para una proteína quinasa que se expresa en músculo esquelético y en otros tejidos y que cumpliría un rol regulatorio: la proteína quinasa de la DM (DMPK), que se muestra en la Figura 4.
Figura 3. Ubicación del gen DMPK en el cromosoma 19 (19q13.3), junto a genes relacionados con otras patologías.
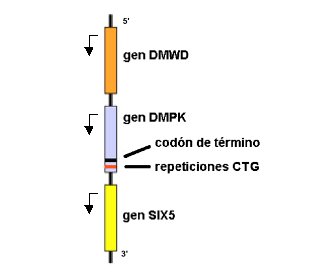
Figura 4. Representación esquemática del locus DM1. La región expandida se localiza en el extremo 3’ no traducido del gen DMPK, pero puede afectar la expresión de los genes vecinos.
En el caso de la DM tipo 1, se produce una expansión inestable de la repetición del triplete CTG ubicada en la región 3’ no codificante del gen DMPK. En los sujetos sanos, este triplete puede repetirse entre 5 y 35 veces, pero cuando ocurre una expansión que alcanza alrededor de 50 veces (estado llamado de premutación), comienzan a manifestarse síntomas leves. Los pacientes más afectados presentan mayor número de repeticiones, las que pueden superar las 2.000 (estado llamado de mutación), como se ilustra en la Figura 5.
Figura 5. Expansión del triplete CTG. En las personas afectadas se encuentra un número aumentado de repeticiones CTG, desde 50 copias en adelante, comparado con 5-37 copias presentes en los individuos normales.
Esta variabilidad se refleja en la gravedad de los síntomas; mientras más repeticiones existan, más afectado se verá el paciente y los síntomas aparecerán más tempranamente en la vida (véase Tabla I). Si el individuo posee más de 50 repeticiones de la secuencia CTG, lo más probable es que desarrolle los síntomas de la enfermedad en algún momento de su vida, aunque en algunos casos, los síntomas serán tan leves que es probable que nunca se haga el diagnóstico de DM.
Tabla I. Correlación entre el número de repeticiones del triplete CTG y la manifestación fenotípica de la enfermedad.
Por lo tanto, la anomalía tiene un efecto pleiotrópico de importancia, ya que su manifestación es multisistémica. Algunos autores afirman que la penetrancia de la enfermedad es muy alta, de casi 100%, mientras que otros describen variaciones que oscilan entre 83% (en Ucrania) y 91% (en Rusia). La expresividad variable se manifiesta en la variación de la gravedad de los síntomas.
Otro aspecto interesante de la DM es el fenómeno conocido como anticipación genética, el cual hace referencia a que el número de repeticiones de la secuencia CTG se irá expandiendo de generación en generación. De este modo, en los descendientes de un individuo afectado la gravedad de los síntomas va aumentando y la edad de aparición de la enfermedad, va disminuyendo, o sea, se presenta cada vez más precozmente.
Existe evidencia de que en hijos que han heredado el gen mutado desde progenitores varones, no ocurre este fenómeno de anticipación genética, que es producido principalmente por un crossing-over desigual durante la profase I de la meiosis, de modo que una de las cromátidas resulta con una ganancia de material genético en la zona de la mutación, aumentando así el número de repeticiones CTG.
El origen de la repetición del triplete CTG en el cromosoma 19, presente tanto en individuos sanos como en individuos enfermos, no ha sido del todo determinado, pero es muy posible que inicialmente se deba a la ocurrencia de una retrotransposición viral. Luego, la expansión de dicha repetición, presente sólo en la enfermedad, se cree que es originada por crossing- over desigual durante la meiosis, que al producirse en generaciones sucesivas causa un aumento del número de repeticiones, generando el fenómeno descrito anteriormente como anticipación genética.
En la actualidad hay consenso sobre la existencia de múltiples factores que contribuyen a la enfermedad. Entre las principales teorías aceptadas para explicar cómo la expansión puede causar DM, está, en primer lugar, la que plantea que la modificación de la cromatina en la región donde se encuentra el trinucleótido repetido altera la expresión de genes adyacentes. En las proximidades de la expansión de la repetición, la cromatina se encuentra condensada de manera anómala, debido a la fuerte cohesión de los nucleosomas, lo que puede resultar en una represión de la transcripción; de hecho, los genes próximos al locus DMPK, DMWD (río arriba) y SIX5 (río abajo) se transcriben menos en presencia de expansiones muy largas de CTG (Figura 4). En modelos experimentales con ratas, se ha demostrado que la no expresión del gen SIX5 produce cataratas.
Una segunda teoría plantea que el secuestro anómalo de proteínas capaces de unirse a secuencias CUG (CUG-BP = CUG binding proteins) lleva a la formación de focos tóxicos de mRNA y a un splicing aberrante. CUG-BP es una ribonucleoproteína nuclear heterogénea (hnRNP), que se une a hebras simples de RNA que poseen la secuencia CUG y que se acumula anormalmente en el núcleo de las células de los pacientes con DM. El rol de las hnRNPs es realizar múltiples funciones regulatorias postranscripcionales tales como el splicing. CUG-BP regula el splicing alternativo y es un sustrato de fosforilación para la DMPK.
Existen dos isoformas de fosforilación, expresadas ubicuamente, tanto en el citoplasma como en el núcleo celular. Es la isoforma hipofosforilada la que se encuentra acumulada en la DM. Esta acumulación afectaría a su vez el splicing de DMPK, generando una DMPK mutante que además de su dominio catalítico, posee dos dominios adicionales: un motivo coiled-coil y una cola que atraviesa la membrana plasmática. Estas isoformas alcanzan pesos de entre 80 kD y 86 kD, lo que influye en la distribución típica de la proteína en los individuos enfermos: agrupada en la membrana de la unión neuromuscular y del retículo sarcoplásmico y aumentada en su relación con la cadena pesada de miosina. Estas numerosas isoformas exhiben especificidades diferentes para distintos sustratos, dependientes tanto del tipo celular, como de la localización intracelular que posean, confiriendo así diferentes roles a la DMPK. Actualmente sólo existen especulaciones sobre el verdadero rol de la DMPK tanto en pacientes sanos como en enfermos.
Se han identificado cinco mRNAs que poseen un splicing alterado en pacientes con DM y cuya aberración podría explicar algunos de los síntomas de la enfermedad: mRNA codificante para tau, proteína relacionada con la miotubularina 1 (MTMR1), mRNA codificante para el receptor de insulina (IR), mRNA codificante para troponina T cardíaca (cTNT) y mRNA codificante para el canal de cloro 1 (ClC-1). La función de MTMR1 en la DM aún se desconoce y la relación entre la proteína tau y alguno de los síntomas aún no ha sido demostrada.
La cTNT humana existe en dos isoformas, que varían en la inclusión o exclusión del exón 5. La que incluye al exón 5 es propia del desarrollo embrionario. Pacientes homocigotos y heterocigotos para DM muestran un aumento significativo de la isoforma que incluye al exón 5.
El receptor de insulina está compuesto por dos subunidades alfa y dos subunidades beta. La ausencia del exón 11 en la subunidad alfa determina la isoforma A (IRA) de ésta y su presencia, la isoforma B (IRB). En la DM la isoforma B, expresada predominantemente en el músculo esquelético, se encuentra principalmente como IRA debido al splicing erróneo de su mRNA.
En la DM, ClC-1 se encuentra en tres isoformas creadas por splicing aberrante, que contienen un codón de término prematuro. Existen mutaciones en canales de sodio y en canales de cloro como ClC-1 que causan miotonía, debido a que se producen defectos en su conductancia. Recientemente, utilizando un modelo de DM en ratas, se determinó que la pérdida de función en este único canal era suficiente para inducir miotonía (Mankodi et al, 2002). La relación entre estos mRNAs (cTNT, IR y ClC-1), es que en todos ellos CUG-BP regula el splicing alternativo uniéndose a regiones ricas un UG en los intrones, en sitios adyacentes a los puntos de splicing.
El mecanismo exacto mediante el cual CUG-BP se acumula en el núcleo de las células en la DM es desconocido. Dos posibilidades se han planteado para explicar estas observaciones: la primera es que la haploinsuficiencia o ausencia total de DMPK favorece la forma hipofosforilada de CUG-BP que se acumula en el núcleo. Esta haploinsuficiencia se debe a que el gen mutado de igual manera se transcribe, pero su mRNA es retenido en el núcleo, así, sólo los transcritos normales son procesados, esto conduce a un déficit de proteína o haploinsuficiencia; además la acumulación de este mRNA tiene efectos citotóxicos. Se sabe que esta retención no se debe ni a interrupción del splicing ni a poliadenilación de los transcritos mutantes.
La segunda explicación sugiere que proteinas de unión a RNAs de doble hebra (dsRNA-BP = double strand RNA binding protein), se unen a loops de la expansión CUG (CTG en el DNA), formando los característicos focos de mRNA e induciendo consecuentemente la elevación de la forma hipofosforilada de la CUG-BP en células con DM.
La DM es la forma más común de distrofia muscular en adultos. El primer caso, documentado por Steinert, Batten y Gibb en 1909, fue descrito como un caso atípico de miotonía congénita. Se estima que 1/20.000 personas tienen la enfermedad a nivel mundial y que en Europa, la proporción es de 1/8.000; sin embargo, algunas poblaciones, por ejemplo, la población residente en la región de Saguenay en Québec (Canadá), poseen una alta incidencia de la enfermedad, de 1/500, debido al “efecto fundador”, que se relaciona con la transmisión de una mutación por un solo individuo que vive en una población muy aislada.
Esta región fue colonizada por los franceses en el siglo XVII y se sabe, gracias a estudios genealógicos, que la mayoría de los casos de DM se originaron de un solo ancestro común. Debido a la inaccesibilidad de esta región hasta avanzado el siglo XX, sufrió pocas migraciones, lo que ayudó a homogeneizar el pool genético de la población y a distribuir ampliamente la mutación.
La DM tipo 2 se presenta en 1/100.000 nacidos vivos.
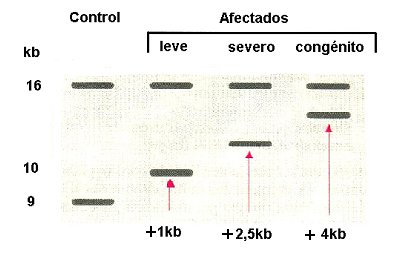
El diagnóstico molecular de la DM se realiza sobre la base del análisis genético, por southern blot, del número de repeticiones del trinucleótido CTG en el gen DMPK. Este análisis del tamańo de fragmentos de DNA que contengan el gen y las repeticiones, permite realizar un diagnóstico. Esta técnica consiste en someter el DNA del paciente a digestiones con enzimas de restricción, seguidas de electroforesis en gel de agarosa junto a un control (normal). Luego, el DNA se transfiere a una membrana de nylon, se denatura y se hibrida con una sonda marcada que posee la secuencia conocida del gen, lo que permite detectar los fragmentos que poseen la secuencia. La migración diferencial del DNA indica el tamańo del fragmento; los fragmentos de mayor tamańo, es decir, aquéllos que contienen mayor número de repeticiones, presentan menor migración.
Existen alrededor de cinco rangos de migración, los que, de mayor a menor, corresponderían a individuos normales, con la premutación, afectados levemente, afectados en forma severa y con la enfermedad congénita. Este último tipo de migración corresponde a la forma más grave de la enfermedad y aparece en aproximadamente 25% de las madres afectadas (Figura 6).
Figura 6. Análisis genético. En las personas afectadas se encuentra un número aumentado de repeticiones CTG (más de 50 copias). Esto se observa en el southern blot como aumento de tamańo (y por lo tanto de la masa) de un fragmento de DNA, es decir, existe una menor migración electroforética (southern blot en locus D19S95, sonda pBBO.7).
También se considera como análisis diagnóstico la reacción en cadena de la polimerasa (PCR = polimerase chain reaction), que permite estimar aproximadamente el número de repeticiones de CTG, siendo más específica que la sola determinación del tamańo de los fragmentos repetidos por el análisis de southern blot (5).
La herencia de la DM es mendeliana, autosómica dominante, pero en los casos en que se presenta el fenómeno de anticipación, en que la secuencia se va repitiendo un número mayor de veces de generación en generación, la gravedad de los síntomas va aumentando, especialmente en el caso de progenitoras mujeres afectadas (8). Si el grado de repeticiones es muy elevado, se puede llegar a manifestar la forma congénita de la enfermedad. Debido a esto, si uno de los padres presenta la enfermedad, es aconsejable hacer las pruebas mencionadas a ambos progenitores para determinar la probabilidad de tener un hijo enfermo. Del mismo modo, también se puede practicar un diagnóstico prenatal, cuantificando el número de repeticiones en el nińo.
En cuanto al manejo, si bien no existe cura para la DM, se recomienda adoptar precauciones para evitar dańo por desconocimiento de la enfermedad. Lo principal es establecer correctamente el diagnóstico y así asegurarse de que los médicos sepan cuando un paciente padece de DM, lo que les permitirá relacionar alteraciones que se presentan en los distintos sistemas orgánicos. Este hecho cobra especial importancia en caso de operaciones y uso de anestesia, ya que en situaciones que impliquen un riesgo vital, como éstas, es necesario tener conocimiento de las alteraciones causadas por la enfermedad a nivel de todos los sistemas, las que podrían provocar cierto grado de descompensación de las funciones vitales.
Se recomienda a los pacientes someterse a controles médicos regulares para manejar posibles complicaciones de la enfermedad, por ejemplo, las de tipo cardiovascular y, finalmente, es importante el cuidado durante el embarazo y el parto, que pueden significar riesgos importantes para la madre.


 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el Curso de Genética, organizado por el Instituto de Ciencias Biomédicas de la Universidad de Chile entre los meses de marzo y julio de 2004.
Edición Científica: Prof. Laura Walker Bozzo, Profesora Encargada de Curso; Prof. Luisa Herrera Cisterna, Coordinadora de Curso.
Expositores:
Lisette Meza G.[1], Catalina Mora L.[1], Daniela Muńoz B.[1]
Citación: Meza L, Mora C, Muńoz D. Myotonic dystrophy: genetic aspects. Medwave 2005 Abr;5(3):e3370 doi: 10.5867/medwave.2005.03.3370
Fecha de publicación: 1/4/2005
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Reardon W, Harley HG, Brook JD, Rundle SA, Crow S, Harper PS, et al. Minimal expression of myotonic dystrophy: a clinical and molecular analysis. J Med Genet. 1992 Nov;29(11):770-3. | CrossRef | PubMed | PMC |Olsen N.K. Myotonic Dystophy. Genetic, neonatologic and neuropsychological aspects. Ugeskr Laeger. 1989 Dec 4;151(49):3300-3. | PubMed |Nesterov LN, Sushcheva GP, Viatkina SIa, Nesterova IM. Myotonic Dystrophy. Zh Nevropatol Psikhiatr Im S S Korsakova. 1983;83(11):1636-41. | PubMed |Ashizawa T, Dubel JR, Dunne PW, Dunne CJ, Fu YH, Pizzuti A, et al. Anticipation in myotonic dystrophy: II. Complex relationships between clinical findings and structure of the CTG repeat. Neurology, 1992; 42: 1877-1883. | PubMed |Wang YH, Amirhaeri S, Kang S, Wells RD, Griffith JD. Preferential nucleosome assembly at DNA triplet repeats from the myotonic dystrophy gene. Science. 1994 Jul 29;265(5172):669-71. | CrossRef | PubMed |Alwazzan M, Newman E, Hamshere M.G, Brook J.D. Myotonic dystrophy is associated with a reduced level of RNA from the DMWD allele adjacent to the expanded repeat. Hum Mol Genet, 1999 Aug; 8 (8):1491-1497. | CrossRef | PubMed |Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem. 2001 Mar 16;276(11):7820-6. | CrossRef | PubMed |Ladd A.N, Charlet N, Cooper T.A. The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol Cell Biol, 2001 Feb; 21(4):1285-1296. | CrossRef | PubMed | PMC |Dunne PW, Ma L, Casey DL, Harati Y, Epstein HF. Localization of myotonic dystrophy protein kinase in skeletal muscle and its alteration with disease. Cell Motil Cytoskeleton. 1996;33(1):52-63. | CrossRef | PubMed |Shimokawa M, Ishiura S, Kameda N, Yamamoto M, Sasagawa N, Saitoh N, et al. Novel isoform of myotonin protein kinase gene product of myotonic dystrophy is localized in the sarcoplasmic reticulum of skeletal muscle. Am J Pathol. 1997 Apr;150(4):1285-95. | PubMed | PMC |Wansink DG, van Herpen RE, Coerwinkel-Driessen MM, Groenen PJ, Hemmings BA, Wieringa B. Alternative splicing controls myotonic dystrophy protein kinase structure, enzymatic activity, and subcellular localization. Mol Cell Biol. 2003 Aug;23(16):5489-501. | CrossRef | PubMed | PMC |Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001 Sep;29(1):40-7. | CrossRef | PubMed |Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998 May 1;280(5364):737-41. | CrossRef | PubMed |






 Estudios originales
Estudios originales