Revista Biomťdica Revisada Por Pares

Este texto completo es la transcripciůn editada y revisada de una conferencia dictada en el curso Lesiones Tumorales y Pseudotumorales del Sistema Musculoesquelťtico, organizado en Santiago por el Departamento de Ortopedia y TraumatologŪa de la Universidad de Chile del 12 al 16 de mayo de 2003.
Director: Dr. Miguel Sepulveda H.
Los tumores osteoarticulares primarios benignos mŠs frecuentes son los tumores formadores de cartŪlago y el osteosarcoma es la neoplasia maligna mŠs frecuente.
El resto son todos tumores poco frecuentes, pero crean problemas tanto en el diagnůstico como en el tratamiento. Algunas de estas entidades no se han podido clasificar en ningķn grupo, porque no concuerdan con sus caracterŪsticas; por eso se agrupan como ďno clasificablesĒ. Son tumores raros y en nuestra experiencia son muy escasos.
Es un tumor maligno por definiciůn, que estŠ limitado al esqueleto axial porque nace de la notocorda. Es de crecimiento lento, de modo que su evoluciůn es tarda e insidiosa. Es poco frecuente.
Se localiza con mayor frecuencia en el crŠneo, aunque en nuestra experiencia la mayorŪa son sacrococcŪgeos y contamos con un par de casos en la columna vertebral lumbar.
En cuanto a las tres localizaciones mŠs importantes del cordoma (crŠneo, vťrtebra y sacrococcŪgeo), hay una diferencia significativa segķn la edad de apariciůn.
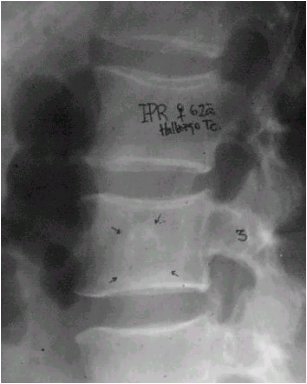
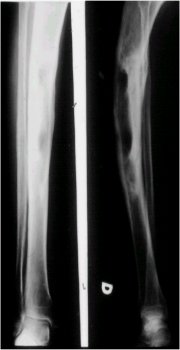
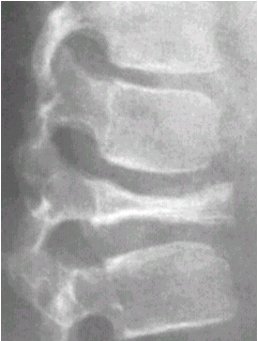
La radiografŪa siguiente muestra una lesiůn en la columna vertebral que no guiaba a un diagnůstico especŪfico. La biopsia realizada en este paciente demostrů que se trataba de un cordoma (Figura 1).
Figura 1. Cordoma vertebral.
Cuando el cordoma se localiza en la columna vertebral, la suma de las localizaciones esfenooccipital y sacrococcŪgea es de 85%, es decir, es un tumor que se localiza en los dos extremos de la columna. Nosotros vemos mŠs la localizaciůn sacrococcŪgea. Se han descrito otras localizaciones, pero son dudosas.
En cuanto a edad y sexo, las lesiones de crŠneo se ven en gente mŠs joven, mientras que las ubicadas en la columna sacra son del adulto mayor. La localizaciůn en vťrtebras no varŪa segķn la edad.
El cordoma es mŠs frecuente en varones y la mayor incidencia de esta lesiůn se encuentra entre los 40 y los 60 aŮos.
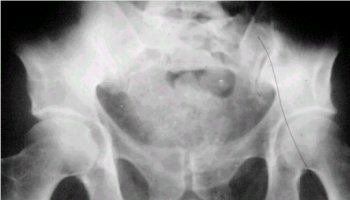
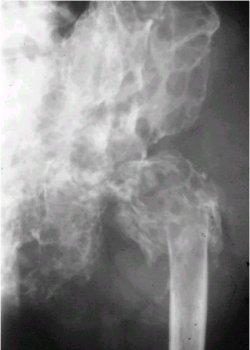
En la radiografŪa de la figura 2 se observa el sacro comprometido en forma completa, con un aspecto algodonoso que es prŠcticamente patognomůnico.
Figura 2. Cordoma en una radiografŪa de pelvis.
El cordoma tiene un comienzo insidioso y sus signos y sŪntomas varŪan segķn su localizaciůn. En el crŠneo se manifiesta precozmente con cefalea, nŠuseas, vůmitos y pťrdida de la visiůn; en cambio, en el sacro los sŪntomas son tardŪos por compresiůn nerviosa: trastornos perineales especŪficos, infecciůn urinaria, alteraciůn de la evacuaciůn o anestesia perineal en silla de montar, segķn la zona que estť comprometida.
En nuestra experiencia, los pacientes con una gran masa antesacra que comprometŪa vejiga y recto presentaban un cuadro clŪnico bastante dramŠtico y de poca soluciůn.
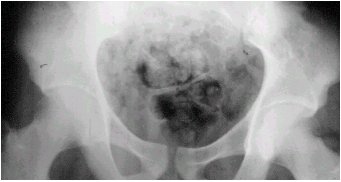
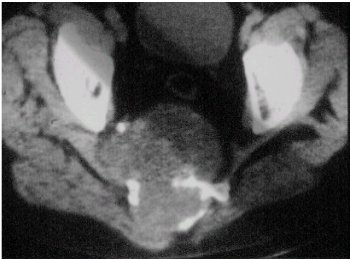
La radiologŪa se puede ilustrar con el caso que se muestra en las figuras 3 y 4, en el que se observan calcificaciones y una lesiůn lŪtica expansiva, de mŠrgenes irregulares, que compromete el sacro.
Figuras 3 y 4. Cordoma del sacro. RadiografŪa y TC.
El cordoma tiene un alto porcentaje de recidiva, porque en general, nunca se reseca el tumor completo. La resecciůn siempre es intralesional o intracapsular, y rara vez es una resecciůn oncolůgica completa.
Sus metŠstasis son tardŪas, debido a la lenta evoluciůn (aŮos) de este tipo de tumor.
Este tumor es mŠs escaso que el anterior; tiene la mitad de su frecuencia. Inicialmente se le denominů adamantinoma de la tibia, pero en la actualidad se denomina ďadamantinoma de huesos largosĒ, porque se ha encontrado localizado en otros huesos. En nuestra experiencia, sůlo lo hemos visto en la tibia.
Tambiťn es un tumor maligno, aunque para algunos no lo es. Es poco frecuente y se ubica en el esqueleto apendicular.
Antes se pensaba que estas lesiones eran secundarias a un golpe directo que causaba una inclusiůn de tejido epitelial hacia las partes blandas, pero en la actualidad se sabe que no existe relaciůn entre golpe y adamantinoma.
Posteriormente, se postulů que la etiologŪa de este tumor era vascular y despuťs se pensů que su origen estaba en el tejido conectivo. Sin embargo, hasta ahora esto no se ha podido determinar.
Es mŠs frecuente en varones, con mayor incidencia en pacientes mayores. Es de evoluciůn lenta y progresiva, causa deformidad y aumento de volumen que, en la tibia, va produciendo una desviaciůn llamada ďtibia en sableĒ.
Evoluciona en muchos aŮos (10 a 20 aŮos), hasta que aparece el dolor, que obliga al paciente a consultar. La localizaciůn mŠs frecuente para nosotros es la tibia, aunque se ha descrito en otros huesos largos (cķbito, fťmur, hķmero, radio).
En los exŠmenes radiolůgicos se puede observar una lesiůn radiolķcida, alargada, bien delimitada, multilocular (de varios tamaŮos), con varias lesiones osteolŪticas, expansiva, que compromete la cortical, la adelgaza y va deformando la diŠfisis, como se puede apreciar en la figura 5.
Figura 5. Adamantinoma de pierna derecha.
No es difŪcil pensar en esta lesiůn frente a un paciente con estas caracterŪsticas; el problema estŠ en que existen otras entidades semejantes. AdemŠs, este tumor tiene un porcentaje importante de recidiva local, da metŠstasis pulmonares tardŪas y se asocia con la displasia fibrosa y la displasia osteofibrosa, lo que complica mucho mŠs el diagnůstico.
Neurilemoma
Es otra entidad rara, menos frecuente que el adamantinoma. Tambiťn se denomina schwanoma o neurinoma. Es un tumor benigno, muy poco frecuente, que compromete el epineuro. No se conoce el mecanismo con que se desarrolla en el hueso.
La mayor incidencia se observa en edades extremas, puede aparecer en niŮos muy pequeŮos (3 aŮos) hasta personas muy adultas (65 aŮos). Es ligeramente mŠs frecuente en mujeres, en las que aparece un peak en la vejez. Es una entidad muy escasa.
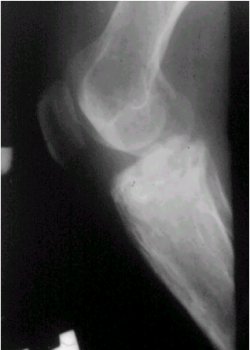
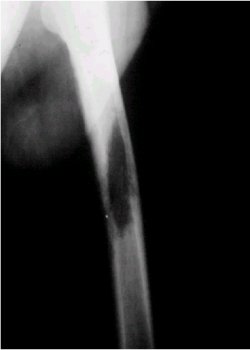
En su presentaciůn clŪnica, se manifiesta con dolor, es de evoluciůn lenta y su localizaciůn es variable. En cuanto a los exŠmenes radiolůgicos, en la figura 6 se puede observar una lesiůn radiolķcida y de ubicaciůn central en la diŠfisis tibial.
Figura 6. RadiografŪa lateral de rodilla que muestra un neurilemoma tibial.
Neurofibroma ůseo
El neurofibroma ůseo, tambiťn raro de ver, es un tumor benigno que en general se asocia con la neurofibromatosis o enfermedad de von Recklinghausen. A nosotros nos interesa mucho mŠs la asociaciůn de ťsta con la escoliosis que con tumor ůseo, pero no se debe olvidar que todos estos tumores pueden tener una forma maligna, que en este caso corresponde al neurofibrosarcoma ůseo.
Son tumores secundarios a enfermedad de Paget, radioterapia, displasia fibrosa y otros cuadros aķn mŠs raros. En nuestra casuŪstica hemos visto algunos casos secundarios a las tres entidades descritas.
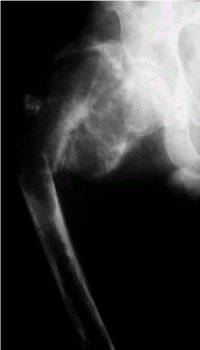
La imagen de la figura 7 corresponde al caso de un paciente a quien se le diagnosticů una enfermedad de Paget mediante biopsia y que despuťs sufriů una transformaciůn hacia osteosarcoma. Se puede ver la gran destrucciůn ůsea.
Figura 7. Osteosarcoma secundario a Paget en cadera izquierda.
En general, sarcomatiza hacia osteosarcoma, pero tambiťn puede hacerlo hacia fibrosarcoma o condrosarcoma. La sarcomatizaciůn secundaria puede afectar cualquier hueso que comprometa la enfermedad de Paget, sobre todo si es mķltiple.
Cuando se utiliza radioterapia en cualquier tumoraciůn ůsea o de partes blandas, se corre el riesgo de una serie de lesiones secundarias. En un paciente joven, se puede daŮar el cartŪlago de crecimiento, provocando trastornos del crecimiento; tambiťn puede provocar necrosis ůsea, que muchas veces es un gran diagnůstico diferencial con metŠstasis, puede producir inflamaciůn o infecciůn ůsea (osteŪtis) y, finalmente, se puede sarcomatizar.
La radioterapia es un sarcomatizador y por eso cada dŪa se utiliza menos. Antes provocaba frecuentemente sarcomas secundarios, que pueden aparecer desde tres hasta treinta aŮos despuťs del tratamiento.
Para definir un sarcoma ůseo como secundario a la radioterapia, cuando llega un paciente que ha recibido radioterapia anteriormente por una patologŪa cualquiera, benigna o maligna, tiene que presentar los cuatro criterios siguientes:
Con respecto a la lesiůn benigna previa, tambiťn puede haber tenido una lesiůn maligna, pero distinta a la posterior.
El mayor temor a esta complicaciůn se debe a lo que ocurriů con el tumor de cťlulas gigantes, que antes se trataba mucho con radioterapia y que en muchos casos presentů sarcomatizaciůn. Sin embargo, este sarcoma es distinto por dos razones: en primer lugar, porque hay mayor probabilidad de que aparezca, despuťs de un tratamiento con radioterapia, en un tumor de cťlulas gigantes que en los demŠs tumores, y luego porque aparece en forma precoz, dos o tres aŮos despuťs de la aplicaciůn de la radioterapia. Por esto, no se debe utilizar radioterapia en estos tumores.
Los tumores secundarios mŠs frecuentes que aparecen despuťs de la radioterapia son: el fibrosarcoma y el osteosarcoma, con las caracterŪsticas ya seŮaladas.
En cuanto a la displasia fibrosa, en la figura 8 se muestra el caso de una paciente portadora de esta lesiůn, que con el curso de los aŮos presentů un cuadro doloroso; se demorů en consultar y, cuando lo hizo, apareciů una sarcomatizaciůn evidente que, en este caso preciso, era un osteosarcoma.
Figura 8. Osteosarcoma en displasia fibrosa previa.
En el aŮo 2000, MartŪnez planteů que la histiocitosis de Langerhans, tambiťn llamada granuloma eosinůfilo o histiocitosis X, era un tumor. Hoy se le reconoce como tal y, en la ķltima ediciůn del libro de la OMS, esta lesiůn aparece descrita como tumor, aunque aķn no estŠ clasificado en forma clara.
Tiene muchos nombres y se presenta en dos formas: solitaria y mķltiple. En la variedad solitaria se encuentra el granuloma eosinůfilo; la variedad mķltiple tiene otras caracterŪsticas y tambiťn estŠ la variedad intermedia, lesiones que no cumplen todos los criterios de las demŠs categorŪas.
Hay variedades crůnicas, agudas y subagudas; las agudas se ven de preferencia en niŮos menores, en los que habitualmente predomina su extensiůn a partes blandas mŠs que al hueso; en los casos crůnicos, en cambio, hay mŠs lesiones ůseas que de partes blandas y se ven en pacientes mŠs adultos.
En nuestra experiencia, la lesiůn tumoral de la histiocitosis X o granuloma eosinůfilo es la misma alteraciůn patolůgica con diferentes manifestaciones clŪnicas, de todo tipo, en otros segmentos y sistemas.
El hueso es una localizaciůn mŠs de esta patologŪa y la forma que mŠs nos interesa es la forma ķnica, conocida como granuloma eosinůfilo solitario. En niŮos, es mŠs frecuente la forma mķltiple; en adultos es mŠs frecuente la solitaria.
Lo mŠs importante es que la mayor mortalidad y las mayores complicaciones ocurren cuando se presenta en edad temprana. En adultos, por lo general, es un hallazgo y muchas veces presenta sŪntomas leves, no como en el primer grupo etario, en el que fallecen mŠs.
En suma, su mayor incidencia estŠ entre uno y quince aŮos de edad, es raro en adultos y es mŠs frecuente en varones, en su forma solitaria.
Cuando la localizaciůn es mķltiple, siempre se debe buscar un compromiso de crŠneo y fťmur. Si se encuentra una lesiůn en el fťmur proximal, se deben buscar dirigidamente otras en el crŠneo. Hay otras entidades que tienen el mismo comportamiento.
La forma solitaria se localiza con mayor frecuencia en crŠneo, costillas, mandŪbula y fťmur. En nuestra experiencia, lo mŠs frecuente es en costillas y fťmur.
Su sintomatologŪa es inespecŪfica, presenta dolor localizado en el segmento de esqueleto afectado, que a menudo duele a la palpaciůn. Tambiťn puede haber aumento de volumen y fractura en hueso patolůgico.
En la radiologŪa, se puede observar una lesiůn radiolķcida bien delimitada, habitualmente con corte en la diŠfisis en la parte proximal del fťmur, como se ve en la figura 9.
Figura 9. Granuloma eosinůfilo en fťmur.
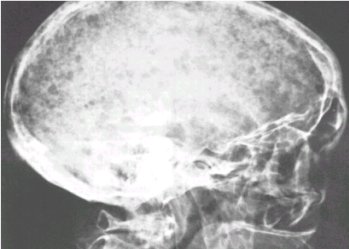
En el crŠneo, presenta lesiones en sacabocado que muchas veces originan confusiůn con otras entidades, como el tumor pardo o el mieloma mķltiple. Tambiťn puede dar aplastamiento vertebral, cuando se localiza a ese nivel; ambas lesiones se pueden ver en las figuras 10 y 11.
Figura 10. Lesiones en sacabocado en el crŠneo.
Figura 11. Aplastamiento vertebral secundario a granuloma eosinůfilo.

 Esta obra de Medwave estŠ bajo una licencia Creative Commons Atribuciůn-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuciůn y reproducciůn del artŪculo en cualquier medio, siempre y cuando se otorgue el crťdito correspondiente al autor del artŪculo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave estŠ bajo una licencia Creative Commons Atribuciůn-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuciůn y reproducciůn del artŪculo en cualquier medio, siempre y cuando se otorgue el crťdito correspondiente al autor del artŪculo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripciůn editada y revisada de una conferencia dictada en el curso Lesiones Tumorales y Pseudotumorales del Sistema Musculoesquelťtico, organizado en Santiago por el Departamento de Ortopedia y TraumatologŪa de la Universidad de Chile del 12 al 16 de mayo de 2003.
Director: Dr. Miguel Sepulveda H.
Expositor:
Miguel Sepķlveda[1]
Citaciůn: Sepķlveda M. Chordoma, adamantinoma, neurogenic tumors, sarcomas after radiotherapy and histiocytosis. Medwave 2004 Ene;4(1):e3300 doi: 10.5867/medwave.2004.01.3300
Fecha de publicaciůn: 1/1/2004
Nos complace que usted tenga interťs en comentar uno de nuestros artŪculos. Su comentario serŠ publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la direcciůn editorial considera que su comentario es: ofensivo en algķn sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas polŪticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisiůn por pares.
Aķn no hay comentarios en este artŪculo.
Para comentar debe iniciar sesiůn







 Estudios originales
Estudios originales