Revista Biomédica Revisada Por Pares

Este texto completo es la transcripción editada y revisada de una conferencia dictada en el curso Lesiones Tumorales y Pseudotumorales del Sistema Musculoesquelético, organizado en Santiago por el Departamento de Ortopedia y Traumatología de la Universidad de Chile del 12 al 16 de mayo de 2003.
Director: Dr. Miguel Sepulveda H.
El grupo de tumores de células redondas comprende el sarcoma de Ewing, el mieloma y el linfoma, que tienen una histología muy similar.
Es una neoplasia maligna de células redondas, pequeńas y azules, que no producen matriz tumoral. Es el segundo tumor más frecuente, después del osteosarcoma.
Afecta a edades muy tempranas y se presenta en menores de 20 ańos; es raro que se presente en mayores de esa edad. La máxima incidencia ocurre entre los 5 y los 15 ańos.
Predomina en hombres, con una proporción de 3:1 respecto a las mujeres. Se caracteriza por su localización en las diáfisis de los huesos largos y en los huesos planos del esqueleto axial (pelvis, escápula, costillas).
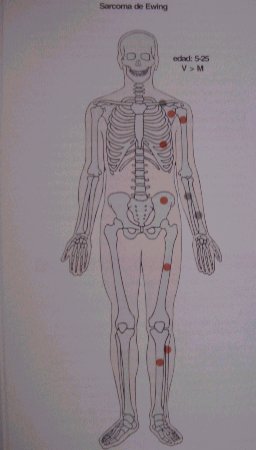
En la figura 1 se aprecia la distribución axial en escápula, costillas, pelvis y en la diáfisis de huesos largos (fémur, tibia, fíbula). Es menos frecuente en ulna y radio.
Figura 1. Distribución más frecuente del sarcoma de Ewing.
Como en todos los tumores óseos, el esquema de abordaje se basa en la anamnesis y el examen físico, además de los exámenes de laboratorio, la radiografía local y de tórax, la cintigrafía ósea, la tomografía computada (TC) local y de tórax, y, por último, en la resonancia nuclear magnética (RM).
Clínica
Suele haber síntomas generales, como fiebre, malestar, pérdida de peso, asociados con la presencia de una masa localizada, dolorosa o con sensibilidad local.
El sarcoma de Ewing tiene dos formas de presentación. En la forma aguda, el cuadro se puede parecer al de la osteomielitis aguda, con la cual se debe hacer el diagnóstico diferencial. La osteomielitis y la artritis séptica son causas frecuentes de consulta por patología ortopédica, en el servicio de urgencia infantil, y se debe tener presente la posibilidad de que en el curso de su estudio aparezca un tumor.
En la forma de presentación clásica, el cuadro se parece al de cualquier otro tumor: el paciente puede consultar por aumento de volumen o por el hallazgo radiológico de una lesión.
El paciente que debuta con un cuadro agudo tiene todos los síntomas de la osteomielitis: fiebre, hemograma alterado con leucocitosis y desviación izquierda, alza de la VHS y de la PCR y anemia. Como la conducta es quirúrgica, se abre el hueso y no se encuentra pus.
El paciente que debuta con un cuadro clásico quizás no tenga un aumento tan importante de la VHS, ni leucocitosis.
Estudio
En un paciente que llega al servicio de urgencia con un cuadro que parece osteomielitis, pero en el que no todo encaja, se debe solicitar radiografía simple, cintigrama óseo, TC local y de tórax, y RM, si se cuenta con un centro de imágenes que funcione las 24 horas, e histopatología. En centros que no cuentan con tales equipos, se debe tomar una decisión sólo con la radiografía y, si se sospecha una osteomielitis, no se puede retrasar la intervención quirúrgica.
En el paciente que debuta con el cuadro clásico se hace todo este estudio antes de llegar a la biopsia.
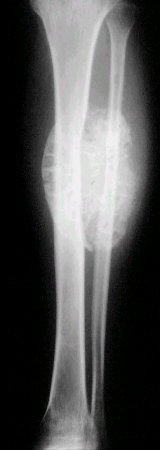
En la figura 2 se observa una lesión diafisiaria de la tibia, con compromiso de la cortical y una gran masa de partes blandas.
Figura 2. Sarcoma de Ewing en la diáfisis de la tibia.
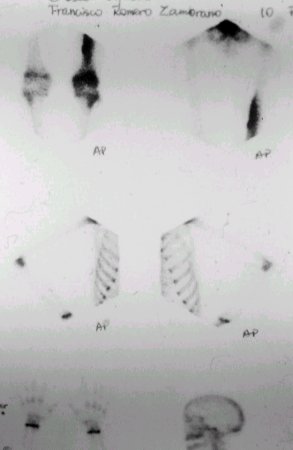
El cintigrama de la figura 3 muestra una lesión del tercio distal del fémur.
Figura 3. Cintigrama óseo con lesión distal de fémur.
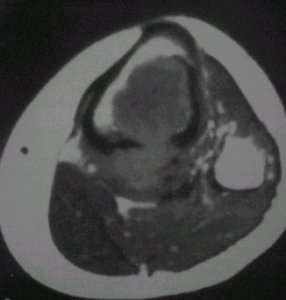
La figura 4 muestra la lesión tomográfica primaria y la masa de partes blandas producida por un tumor de Ewing.
Una diferencia característica, entre el osteosarcoma y el sarcoma de Ewing, es que en éste la lesión se acompańa de un aumento importante de las partes blandas en la TC y la radiografía, lo que no ocurre en el primero.
Figura 4. Tumor de Ewing en la TC.
En la RM de la figura 5 se puede ver el compromiso medular y la masa de partes blandas.
Figura 5. Tumor de Ewing: resonancia magnética de muslo.
Diagnóstico diferencial
Las principales entidades que se pueden confundir con este tumor son:
La biopsia puede ser abierta o cerrada.
Tratamiento
Consiste en quimioterapia, radioterapia y cirugía.
Aún se discute si es un tumor óseo o un tumor secundario; sin embargo, aunque algunos consideran que la médula no es tejido óseo, en la clasificación de la Organización Mundial de la Salud (OMS) figura como tumor óseo.
Es un tumor maligno que se origina en la médula ósea por una proliferación de las células plasmáticas. Este tumor, según el servicio donde se realicen los estudios, puede competir con las metástasis, en cuanto a frecuencia.
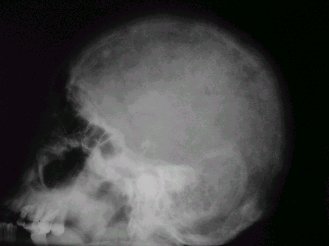
En el mieloma múltiple, la radiografía puede mostrar imágenes en sacabocado, tal como se aprecia en la figura 6.
Figura 6. Radiografía lateral de cráneo.
El mieloma solitario o plasmocitoma es igual al mieloma múltiple en su histología, pero se ve en edades más tempranas y se considera que no es más que una fase precoz de éste.
En cuanto a epidemiología, el mieloma múltiple es el segundo tumor óseo más frecuente después de las metástasis, y corresponde a 17,6 % de los tumores óseos malignos. Se presenta sobre los 40 ańos, entre la quinta y la séptima década de la vida, afecta de manera predominante al sexo masculino (3:1) y se localiza preferentemente en el esqueleto axial.
Clínica
Una forma de presentación frecuente es el dolor lumbar importante; el aumento de volumen es raro y sólo 10% de los casos debutan con fractura. Hay síntomas generales, como fiebre, baja de peso y compromiso del estado general. El paciente se ve comprometido, con dolor.
Laboratorio
En el hemograma presenta una anemia normocítica y normocrómica. La calcemia está aumentada y la función renal va a estar comprometida, en las fases más tardías, por una amiloidosis renal.
La inmunoelectroforesis de proteínas es importante en el diagnóstico: se observa un peak monoclonal, generalmente de la IgG (65 %). Hay una inversión de la relación albúmina/globulina.
El estudio se realiza con radiografía simple, cintigrafía, laboratorio, mielograma y biopsia, en la cual se basa alrededor de 27% de los diagnósticos.
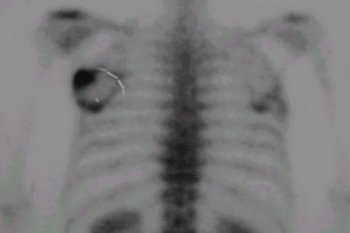
En el cintigrama de la figura 7 se ve un halo hipocaptante en relación al tumor. Se debe pensar en un mieloma cuando la periferia es hipocaptante y el centro, muy hipercaptante.
Figura 7. Mieloma múltiple: cintigrama óseo.
El diagnóstico diferencial se plantea con las metástasis (pelvis, vértebras, múltiples), la osteoporosis, el tumor pardo del hiperparatiroidismo y el linfoma.
Etapificación
En el mieloma múltiple se utiliza la etapificación de Durie/Salmon, que se basa en la carga tumoral del proceso y tiene aspectos pronósticos importantes. Para asignar la etapa, se miden las concentraciones séricas de calcio, hemoglobina y paraproteína, y se determina la excreción urinaria de la proteína de Bence Jones y el número de lesiones esqueléticas.
Con esto se ubica al tumor en uno de los tres estadíos (I, II y III). Siempre intervenimos en el estadío II cuando ya hay compromiso óseo.
Los factores que determinan el pronóstico son la carga tumoral, la insuficiencia renal (creatinina mayor de 2 mg%), el nivel de albúmina menor de 3 gr (por peak de inmunoglobulinas) y la proteína C reactiva mayor a 0,4 mg%.
La sobrevida media sin tratamiento es menor de un ańo. Con tratamiento de melfalan prednisona llega a dos a tres ańos.
Está el linfoma primario (Hodgkin y no Hodgkin) y el linfoma secundario, que es el más frecuente.
El linfoma óseo primario, también denominado reticulosarcoma o linfosarcoma, es muy infrecuente (3-4%). Para catalogarlo como primario no debe existir otra localización extraósea ni ganglionar. Hay 30% de compromiso óseo en los linfomas malignos.
En el linfoma secundario, el compromiso óseo es secundario a un linfoma nodal o extranodal que afecta el hueso por continuidad o por diseminación hematógena.
Presentación clínica
Si bien se observa en pacientes entre los 20 y los 70 ańos, la máxima incidencia se observa entre los 50 y los 70, con una proporción de hombres a mujeres de 3:2.
En 25% de los casos debuta con fracturas y se localiza, por lo general, en huesos largos, pelvis, costillas y vértebras. Los síntomas locales son muy similares a los de otros tumores: dolor, aumento de volumen y masa palpable, según la localización.
Se puede presentar con síntomas sistémicos, como fiebre, pérdida de peso, ictericia, etc., pero también puede ser asintomático y diagnosticarse como un hallazgo.
Estudio
Se realiza con radiografía convencional, cintigrama óseo, TC local, TC de tórax, abdomen y pelvis, RM e histología.
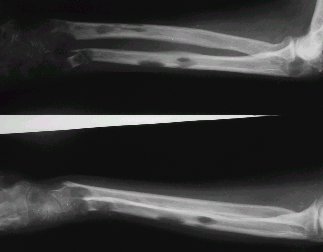
En la radiografía de la figura 8 se observan imágenes líticas múltiples que van hacia la cortical, excéntricas, ubicadas a lo largo del hueso.
Figura 8. Linfoma óseo. Radiografía convencional de antebrazo.
Entre los linfomas óseos, el más frecuente en el linfoma no Hodgkin. El compromiso óseo del linfoma de Hodgkin es muy poco frecuente.
Linfoma de Hodgkin óseo
Es extremadamente raro. Afecta principalmente las vértebras y aparece como una lesión muy condensante y esclerosante. Generalmente, la diseminación es desde un foco extraóseo (5 a 21%).
En cuanto a su presentación clínica, generalmente produce un dolor que precede a la imagen radiológica. Ocasionalmente, hay aumento de volumen en la zona de la columna sobre el hueso afectado.
Entre las complicaciones que origina están las fracturas espontáneas, en 25% de los casos, la compresión medular o radicular en la localización vertebral y la hipercalcemia.
El diagnóstico diferencial se plantea con sarcoma de Ewing, osteosarcoma, metástasis, plasmocitoma, osteomielitis, enfermedad de Paget y neuroblastoma metastásico.

 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de una conferencia dictada en el curso Lesiones Tumorales y Pseudotumorales del Sistema Musculoesquelético, organizado en Santiago por el Departamento de Ortopedia y Traumatología de la Universidad de Chile del 12 al 16 de mayo de 2003.
Director: Dr. Miguel Sepulveda H.
Expositor:
Orlando Wevar[1]
Citación: Wevar O. Round cell tumors: clinical presentation. Medwave 2003 Dic;3(11):e3282 doi: 10.5867/medwave.2003.11.3282
Fecha de publicación: 1/12/2003
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión







 Estudios originales
Estudios originales