Revista Biomédica Revisada Por Pares

Este texto completo es la trancripción editada y revisada de la conferencia dictada en la XIV Reunión Científica de la Sociedad Interamericana de Hipertensión, 25-29 de marzo de 2001, Santiago.
Editora Científica: Dra. Gloria Valdés.
Introducción
En esta presentación se mostrará lo que actualmente se está realizando, con relación al papel del óxido nítrico (NO) en la regulación de la respiración celular, y cómo puede llegar a tener un papel fisiopatológico basado en su importante acción fisiológica en el sistema cardiovascular.
Situaciones donde el NO desempeńa un papel fisiopatológico
Desde el inicio de la investigación del NO, ha llamado la atención que la “Vía de L-arginina-NO”, como se la ha denominado, tiene funciones fisiológicas significativas, no solamente en el sistema cardiovascular sino también en el sistema nervioso central y periférico, en la inmunología y en la inflamación.
En cada uno de estos sistemas biológicos, el NO puede transformarse, bajo ciertas condiciones, en una entidad importante en situaciones fisiopatológicas. Por ejemplo, en el sistema cardiovascular, la producción excesiva de NO es importante en la producción de hipotensión en el shock séptico y, probablemente, en el dańo tisular secundario. A nivel del sistema nervioso central, donde el NO es muy importante como mediador fisiológico, un exceso en su producción puede ser importante en el desarrollo de enfermedades neurodegenerativas.
Esto lleva a preguntarse en qué condiciones el NO, de ser una entidad fisiológica, se transforma en una fisiopatológica. Una de ellas tiene que ver con la función normal y la otra, con la enzima que sintetiza NO. Esta es la enzima inducible, a diferencia de la endotelial y la neuronal, que son enzimas constitutivas que generan pequeńas cantidades de NO como mecanismos de seńal. Cuando los glóbulos blancos, que no producen en forma constitutiva el NO, son activados por lipopolisacáridos (LPS) o ciertas citoquinas, existe una inducción de novo de NO inducible. Este mecanismo de defensa del organismo contra microorganismos invasores puede ser la base de algunas de estas situaciones, en que la producción excesiva de NO durante un tiempo largo produce dańo tisular del huésped. No existe ninguna duda de que el aumento de la producción de NO explica la hiporreactividad y la hipotensión del shock séptico y cada día se acumula más evidencia de que algunos de los dańos tisulares del shock séptico se deben a la producción de NO. Por otra parte, está claro que la producción aumentada de NO tiene un papel importante en la inflamación crónica reagudizada y en ciertas enfermedades degenerativas.
El NO por sí solo, probablemente, no es tóxico. Aun después de períodos largos, el NO no produce dańo tisular por sí solo, sino que necesita interactuar con ciertas moléculas. Un ańo antes de la identificación del EDRF como NO, junto a Gryglewsky y Palmer, demostramos que los iones superóxido participaban en la degradación del EDRF (Proc Natl Acad Sci U S A 1986c;83:9164-8). Este hallazgo fue muy significativo al ańo siguiente, tanto en nuestros experimentos como en los de Furchgott, para la identificación del NO y para comprender de qué manera el NO podría participar en el dańo tisular.
Interacción entre superóxido y NO
En un bioanálisis farmacológico muy hermoso realizado en 1986 se demostró cómo la superóxido dismutasa (SOD) dismuta el superóxido y lo saca del medio, con lo cual consigue aumentar, de la misma manera, la vida media del EDRF liberado por bradicinina y la vida media del NO auténtico.
En el modelo de vasos en serie se demuestra que hay una liberación de EDRF que desaparece rápidamente en la cascada, comparado con el NO auténtico que desaparece exactamente con la misma rapidez. En presencia de una infusión de SOD, la vida media de ambos compuestos aumenta exactamente de la misma manera y, cuando se termina la SOD, nuevamente retorna a la vida media original. Esto demostró que el superóxido es importante para destruir el NO.
Un ańo más tarde, trabajando con Therese McCall, estudiante de doctorado de nuestro laboratorio, encontramos que, si se activan glóbulos blancos, ellos liberarán superóxido y NO, y la interacción de ambos anula sus actividades biológicas. No estaba claro lo que ocurriría si estas dos sustancias interactuaban, pero se plantearon dos posibilidades: que interactuaban para producir nitrato, con una toxicidad reducida, o que interactuaban para producir fracciones más tóxicas. Al ańo siguiente, Beckmann encontró que el NO y el superóxido interactúan para producir peroxinitrato, especie muy oxidativa que al ser protonada forma radicales hidroxi y NO2. Desde entonces, la producción de peroxinitrato ha sido investigada en muchas situaciones patológicas distintas, incluso la hipertensión y la aterosclerosis. Además, la formación de peroxinitrato, medida indirectamente por la nitrosilación de proteínas, produce una reacción tipo nitrosilación que se ha identificado en estas condiciones.
Relación entre peroxinitrato y NO
Uno de los campos más interesantes en la investigación del NO es la interacción y la producción de peroxinitrato, y las condiciones en las cuales éste puede causar dańo. Al parecer, no siempre que se produce peroxinitrato ocurre dańo tisular. Hace algunos ańos se observó que existen mecanismos eficientes de barrido o limpieza, en los tejidos, que inactivan el peroxinitrato. El mecanismo más fuerte se compone de tioles con los cuales el peroxinitrato interactúa para producir nitroso-tioles, que regenerarán NO. En esa situación no habrá dańo tisular; en cambio, en situaciones de menor concentración de tioles, el peroxinitrato sí podría causar dańo.
Esto es muy importante, porque permite plantear que el destino y la acción del peroxinitrilo depende, en gran parte, del ambiente biológico en que se produce y que los trabajos deben orientarse a dilucidar en qué condiciones es capaz de producir dańo. Por ejemplo, se sabe que la generación simultánea de superóxido y NO, que ocurre a nivel de la placa de aterosclerosis, puede iniciar la peroxidación de lípidos como, por ejemplo, las lipoproteínas de baja densidad. Por lo tanto, es muy crítico saber cuándo se va a producir peroxinitrato y en qué condiciones va a actuar como un oxidante potente.
A partir de esto, se planteó que la producción de peroxinitrato, en un microambiente determinado, puede ser más dańina que si se produce en forma más general, ya que en este caso se puede barrer más fácilmente. Lo dicho originó todo el trabajo realizado en los últimos ańos, con el objeto de determinar cómo el NO actúa como entidad patológica a nivel de la mitocondria.
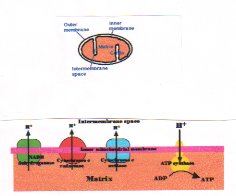
Es muy importante recordar que el NO,en concentraciones fisiológicas, activa la guanilatociclasa soluble, también inhibe esta enzima. En la cadena de la fosforilación oxidativa de la mitocondria existen tres complejos: complejo I, II y III (Figura 1). El NO y el oxígeno compiten por estas enzimas, lo cual es muy importante, porque el proceso ocurre con una concentración fisiológica de oxígeno y NO. Esto condujo a pensar que el NO seguramente es un regulador fisiológico de la respiración celular. Es importante recordar nuevamente que guanilato ciclasa y las enzimas de la cadena respiratoria son las únicas enzimas sobre las cuales el NO actúa fisiológicamente.
Figura 1.
El NO como regulador de la respiración celular
Para investigar este aspecto se colocaron células endoteliales dentro de una cámara de oxígeno cerrada, para ver cómo consumían este gas. Con esto se pudo confirmar una observación hecha muchos ańos antes: que si se ponen células a respirar en una cámara de oxígeno cerrada, para que se reproduzcan, el consumo de oxígeno no es lineal. Cuanto más bajo el oxígeno, menos lo consumen las células, porque existen mecanismos de regulación que funcionan según las concentraciones de oxígeno tisular.
También se observó que cada vez que se administraba bradicina y se liberaba NO, se hacía más lenta la respiración celular. Pero lo más interesante fue la forma de la curva, que mostraba cómo disminuía el consumo de oxígeno a medida que bajaba su concentración. Entonces se trataron las células con un inhibidor de la NO sintasa y se encontró que, en ausencia de NO, las células respiraban en forma constante a lo largo de toda la curva de concentración de oxígeno, algo muy distinto de lo que ocurría con las células de control.
Esto indica que, a medida que la concentración de oxígeno baja, se produce una disminución de la respiración, debido a que el NO y el oxígeno compiten, y a que la importancia relativa del NO aumenta. En condiciones sin NO, la bradicinina no disminuye la respiración, porque no hay liberación de NO.
En un experimento realizado hace tres o cuatro ańos, se concluyó que la razón NO/oxígeno modula la actividad de la citocromo-c-oxidasa, lo que confiere a esta enzima la capacidad de ser un sensor de oxígeno en las células, ya que, según la concentración de NO, la célula detectará altas o bajas concentraciones de oxígeno. Esto es importante, porque durante muchos ańos se han buscado sensores de oxígeno y este mecanismo representa un regulador importante y simple de las cantidades de oxígeno que consumirá la célula en distintas situaciones metabólicas.
En lo que respecta al dańo tisular, es importante saber qué ocurre si el NO aumenta en una célula a tal punto que no haya suficiente oxígeno para desplazarlo. Esto es bastante posible, porque la concentración de oxígeno, a nivel de la mitocondria, es de alrededor de ocho a diez micromolar y la concentración de NO descrita es muy cercana a la que interactúa para producir IC-50; luego es muy probable que bajo ciertas condiciones el NO aumente hasta un punto en que no hay suficiente oxígeno para desplazarlo. En tales condiciones, el NO se comporta como un inhibidor metabólico y los únicos otros dos inhibidores de estas enzimas, que son bien conocidos y ambos irreversibles, son el monóxido de carbono y el cianuro. Por lo tanto, en el cuerpo existe una molécula reguladora muy potente fisiológicamente, que puede comportarse como un inhibidor metabólico al aumentar las cantidades de NO y, en tales condiciones, se llegó a la conclusión de que es probable que el superóxido se genere en la fase de óxido-reducción de la cadena de la fosforilación oxidativa, con lo cual se genera peroxinitrato dentro de la mitocondria y se inicia el dańo tisular. Esta secuencia de episodios llevaría de una regulación fisiológica a una patológica, porque ya se ha demostrado que el peroxinitrato, diferente de NO, produce dańo irreversible en algunos complejos mitocondriales. Esta es la idea con que estamos trabajando hace varios ańos.
Se ha encontrado bastante evidencia a favor de esta hipótesis. Se ha visto que si se inhibe la citocromo-c-oxidasa durante tiempos largos, el NO celular produce consumo de glutatión de las células. El glutatión es el principal mecanismo antioxidante de la célula. Hemos encontrado que cuando el glutatión disminuye, el NO o el peroxinitrato, probablemente, inhiben el complejo I de manera irreversible, mediante la nitrolización de tioles. Entonces, la secuencia de episodios parece partir de una inhibición del complejo IV, la generación de superóxido en la cadena de óxido reducción, la generación de peroxinitrato, la interacción de tioles, incluso algunos tioles principales en las células, como el complejo I y probablemente otras enzimas.
Papel del NO en la muerte celular
żQué pasa en los primeros segundos o minutos cuando el NO actúa sobre estas enzimas? żCómo responde la célula cuando percibe que la citocromo-c-oxidasa está inhibida?
La literatura sobre NO y sobrevida o muerte celular se divide en dos grandes grupos: uno, alrededor de 50%, que cree que el NO protege las células, y otro 50% que cree que el NO causa la muerte celular. Estos grupos no tienen la posibilidad de dialogar entre ellos.
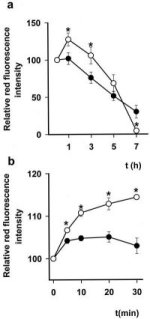
En los últimos meses se han obtenido resultados muy interesantes con sólo inhibir la respiración celular, usando NO, y observar lo que ocurre. Se midió la sobrevida celular usando un método de deprivación de suero y buscando la aparición de apoptosis en un plazo de 14 horas. La apoptosis apareció lentamente y lo más sorprendente fue que las células sin NO fueron protegidas en las primeras horas y luego murieron súbitamente, con mayor rapidez que las controles, lo que coincide con el tiempo en que el glutatión disminuye y se produce el estrés oxidativo (Figura 2).
Figura 2.
También se ha descrito en la literatura que, si se administra NO a las células, el potencial de membrana mitocondrial se despolariza y se inicia el dańo. Con esta base, se hizo exactamente el mismo experimento, midiendo el potencial de la membrana mitocondrial. Nuevamente se encontró que en las primeras horas el NO aumentaba el potencial de la membrana mitocondrial y que en ese lapso las células estaban protegidas del dańo, y que sólo después, cuando ya había estrés oxidativo, las células disminuían su potencial de membrana. El control, a los 30 primeros minutos, mostraba claramente que el potencial de membrana aumentaba, en abierta contradicción con lo descrito en la mayor parte de la literatura. Por esto, se decidió medir el potencial de membrana con microscopía confocal. Se encontró que agregar NO no inducía la polarización de las mitocondrias; la mitocondria aumenta el potencial de membrana inmediatamente, a medida que se inhibe la respiración, y durante un tiempo largo la célula está protegida contra la muerte, porque el potencial de membrana de la mitocondria está aumentado (Figura 3).
Hipótesis actual
Nuestra hipótesis actual se basa en información que está en vías de publicación. En la fosforilación oxidativa de la célula existe la vía glicolítica, que produce piruvato, el cual cambia a ácido cítrico para producir NADH, el que va a la vía de la fosforilación oxidativa (Figura 5 =cadena de fosforilacion). La fosforilación oxidativa está controlada en el último paso por el NO. A medida que los electrones pasan a lo largo de la vía empujan a los protones, los que van al espacio intermembrana de la mitocondria y luego regresan al citoplasma mitocondrial (Figura 1 A), activando la ATPasa para que genere ATP. Este es el control y regulación fisiológicos de la fosforilación oxidativa acoplada a la síntesis de ATP.
Nuestra hipótesis plantea que, cuando aumenta el NO, la célula lo percibe como una disminución de oxígeno e inmediatamente las células que tienen la capacidad de usar glucosa, cambiándose a glicólisis, revierten la función de la ATPasa y utilizan el ATP, para mantener el potencial de membrana. Esta es la fase protectora de la acción del NO. Sin embargo, a medida que ocurre esta inhibición, la glicólisis empieza a desaparecer por falta de sustrato. Probablemente, el dańo mitocondrial ocurre por cambios de pH. Existe un estrés oxidativo, como ya se ha mencionado, y se produce S-nitrosilación de proteínas, salida de calcio, despolarización de la mitocondria, liberación de citocromo-c y finalmente, muerte celular. Es muy probable que estas sean las etapas, porque ya existe evidencia de que el citocromo-c no sale de la mitocondria al tiempo que aumenta el potencial de membrana, así que seguramente el NO no es sólo un mecanismo protector, sino que además es algo contra lo cual las células se autoprotegen de manera eficaz en las primeras etapas.
Esta hipótesis ya está planteada y, probablemente, cuando se comprenda la regulación fisiológica de la respiración celular, se podrá entender cómo la inhibición de la respiración lleva a esta cadena de hechos que abarca importantes cambios en el pH intracelular, estrés oxidativo, S-nitrosilación, cambios en el potencial mitocondrial y dańo celular final.
Conclusión
Estos estudios podrán ayudar a entender la sobrevida celular, la apoptosis y la necrosis.
Uno de los hechos que más entusiasma en esta área es la posibilidad de que la NO sintasa esté presente en la membrana interna de la mitocondria. En un estudio recientemente publicado, que enfocó la producción de NO en células, mediante un equipo especial (mitocondria tracker) también se observaron mitocondrias que respiraban activamente; luego se realizó fluorescencia combinada y se encontró que la mayor parte de la fluorescencia de NO está dentro de la mitocondria. Si esto es realmente así y la NO sintasa está presente en todas las mitocondrias del cuerpo, entonces sería de gran importancia en los mecanismos reguladores fisiológicos, porque estaría lista para producir el dańo si la secuencia de hechos que se ha descrito es efectiva.

 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la trancripción editada y revisada de la conferencia dictada en la XIV Reunión Científica de la Sociedad Interamericana de Hipertensión, 25-29 de marzo de 2001, Santiago.
Editora Científica: Dra. Gloria Valdés.
Expositor:
Salvador Moncada[1]
Citación: Moncada S. Nitric oxide: role of NO in cellular respiration. Medwave 2001 Oct;1(01):e764 doi: 10.5867/medwave.2001.10.764
Fecha de publicación: 1/10/2001
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión







 Estudios originales
Estudios originales