Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Este texto completo es la transcripción editada y revisada de la conferencia dictada en el marco del Simposio Hiperglicemia Postprandial, Factor Emergente de Riesgo Cardiovascular en la Diabetes Tipo 2, realizado el 11 de abril de 2002. El evento fue organizado por Novartis.
Es curioso que se hable de la importancia de la hiperglicemia postprandial como un concepto emergente, porque la diabetes es de hecho una hiperglicemia postprandial y se define por la presencia de un alza glicémica después de una comida.
La razón por la que se ha vuelto a plantear este concepto es que hace un tiempo los colegas de Norteamérica estimaron que no tenían dinero suficiente para realizar pruebas orales de tolerancia a la glucosa en sus pacientes para diagnosticar la diabetes, y decidieron reemplazarlas por reglas para definir esta enfermedad, las que, por supuesto, debían ser válidas en todo el mundo, basadas en los niveles de la glucosa plasmática en ayunas. Incluso inventaron una nueva enfermedad llamada “impaired fasting glicemia”, o glicemia en ayunas deteriorada, para ocultar el hecho de que estaban simplemente tratando de ahorrar dinero.
Éste fue un gran error, porque la diabetes es de hecho una enfermedad de una etapa de la alimentación. Si se mantiene en ayunas a cualquier paciente con diabetes tipo 2 su glicemia disminuirá y finalmente se normalizará, y volverá a aumentar sólo cuando se alimente nuevamente.
Ahora la Asociación Americana de Diabetes está intentando enmendar sus errores y volver sobre sus pasos, para lo cual pretende introducir otra vez el concepto de que, para documentar la existencia, la gravedad y el pronóstico de la diabetes, es necesario medir los niveles de glucosa posterior a una ingesta de glucosa o de una alimentación mixta.
Una vez aclarada esta confusión sobre un “nuevo” concepto, que en realidad no es nuevo para nada, es preciso repasar el concepto de hiperglicemia postprandial.
Al medir la glucosa plasmática y la concentración de insulina en un grupo de pacientes con diabetes tipo 2 y en un grupo control de sujetos no diabéticos, durante 15 horas de vida normal en las que se incluye el desayuno, el almuerzo y la comida, se observa que existe un estrecho control de la glucosa en los individuos no diabéticos (Figura 1). Es importante recordar que la concentración de glucosa plasmática es una variable homeostásica muy controlada, que se mantiene dentro de un rango de valores muy estrecho, y que esto se consigue gracias a la respuesta insulínica, que se presenta rápidamente después de una ingesta.
En cambio, la diabetes se caracteriza por una pérdida completa de la homeostasis de la glucosa, que se traduce en una hiperglicemia en ayunas, de modo que los diabéticos tipo 2 típicos, con un índice de masa corporal (IMC) de alrededor de 28, tienen niveles de glucosa plasmática en ayunas dos veces más altos que los individuos no diabéticos, y sus alzas glucémicas postprandiales son también mucho mayores.
Por lo tanto, la diabetes se define por la presencia de hiperglicemia en ayunas y postprandial, y esta información se puede reproducir en cualquier clínica u hospital. Mientras más completo es el ayuno, más amplia es la excursión (o alza) de la glicemia postprandial.
Sin embargo, no es fácil documentar cuál de los dos estados, el de ayuno o el postprandial, es causa del aumento general del nivel de glucosa plasmática en 24 horas, porque esto depende de la gravedad de la diabetes. Cuando los niveles de glucosa plasmática en ayunas son medianamente elevados, la contribución de la hiperglicemia en ayunas no es tan importante, pero, cuando estos niveles son más elevados, la excursión es mayor y por lo tanto la contribución de la hiperglicemia postprandial se hace más importante.
Entonces, el debate que aparece en la literatura sobre cuál de las dos hiperglicemias, la de ayuno o la postprandial, contribuye más a la glicemia total, está un poco fuera de lugar, ya que depende de la gravedad del cuadro. La mayoría de las veces, en los diabéticos, el nivel de glucosa plasmática está por sobre el valor normal en ayunas porque aquéllos no logran regresar a su nivel glicémico basal en forma rápida, como los no diabéticos.
Si se aplica el concepto epidemiológico de exposición, que es el producto del aumento del nivel de glucosa por el tiempo durante el cual existe este aumento, y si se acepta que esta exposición es biológicamente dañina para los órganos vitales, se puede concluir que el daño terminal de órganos en el diabético es consecuencia de los niveles elevados de glucosa en ayunas y postprandial.
Patrones de hipo e hiperinsulinemia en diabéticos tipo 2
Los niveles plasmáticos de insulina en los individuos diabéticos tienen perfiles diferentes de los observados en individuos no diabéticos, pero no están reducidos en términos absolutos. Observando los cambios en los niveles de glucosa plasmática, en relación con la respuesta de insulina plasmática en individuos diabéticos y no diabéticos, se puede apreciar que el déficit no es tan importante en promedio en la diabetes, porque hay períodos con niveles bajos de insulina, pero éstos aumentan mucho entre las comidas (Figura 2).
Por lo tanto, los pacientes diabéticos alternan los estados de hipoinsulinemia y de hiperinsulinemia, y por eso se dice que los dos grandes defectos de los diabéticos son la insulinorresistencia o hiperinsulinemia, y la hipoinsulinemia. Ninguno de los dos es exclusivo, pero sí lo es la existencia simultánea de estos dos estados en un paciente.
Impacto de la hiperglicemia de ayunas y postprandial
Numerosos estudios han analizado el efecto de la hiperglicemia en ayunas y de la hiperglicemia postprandial sobre los órganos, pero es importante mencionar dos de ellos. En primer lugar está el DECODE (Diabetes Epidemiology: Collaborative Analysis of Diagnostic Criteria in Europe), cuyos resultados fueron publicados hace dos años en The Lancet. En él se analizaron datos procedentes de 13 estudios de cohorte, prospectivos, de Europa, que abarcaban, aproximadamente, a 25.000 pacientes, y se determinó el riesgo de muerte según las diferentes categorías diagnósticas de la glucosa en un seguimiento promedio de 7 años (1).
Al analizar el riesgo relativo de mortalidad por todas las causas, según los niveles de glucosa plasmática en ayunas y a las dos horas de administrar una carga de glucosa, se observó que los niveles de glucosa a las dos horas aumentaban más el riesgo que el incremento de los niveles de glucosa en ayunas, aunque estos resultados no son postprandiales, sino solamente postcarga de glucosa (Figura 3).
Con esta información, se calculó el riesgo relativo cardiovascular, de enfermedad coronaria, de accidentes vasculares encefálicos y de mortalidad por todas las causas, en pacientes diabéticos asintomáticos, pues el riesgo sería muy distinto si el paciente ya fuera sintomático de enfermedades cardiovasculares.
Se observó que la glicemia en ayunas mayor de siete milimoles, según la definición de la Asociación Americana de Diabetes, aumentaba el riesgo relativo de cada uno de estos episodios; pero al hacer un ajuste simultáneo respecto a la concentración de glucosa a las dos horas, sólo la mortalidad por todas las causas mostraba un riego aumentado, con una importancia estadística limítrofe(2), como se puede ver en la Tabla I:
Tabla I. Riesgo relativo* de mortalidad en pacientes diabéticos asintomáticos según FPG (ADA).
También se ajustó respecto a los predictores de riesgo comunes de enfermedad cardiovascular: edad, sexo, colesterol, presión arterial, tabaco e IMC.
En otras palabras, si se analizaba el grupo clásico de factores de riesgo y se calculaba el mayor riesgo debido a una glicemia en ayunas superior a seis milimoles por litro, resultaba importante, pero, cuando se agregaba al modelo la glicemia a las dos horas, ya no lo era.
En otro estudio, Marcos Hanefield analizó la incidencia anual de infartos al miocardio y la mortalidad según niveles de glucosa en ayunas y postprandial, sin niveles postglucosa, y observó una tendencia al aumento de los infartos y la mortalidad, a medida que aumentaba la glicemia en ayunas, y un aumento más definido y estadísticamente importante con los niveles aumentados de glicemia postprandial(3).
Con los resultados de estos dos estudios epidemiológicos longitudinales, se determinó que la glicemia posterior a una carga de glucosa, o glicemia postprandial, es tan importante o más que la glicemia en ayunas para predecir el riesgo cardiovascular en pacientes diabéticos tipo 2. No todos los estudios disponibles concuerdan, pero la evidencia favorece este concepto, a tal punto que la Asociación Americana de Diabetes ya aceptó incluir la hiperglicemia postprandial dentro del grupo de factores de riesgo cardiovascular en pacientes diabéticos.
Las conclusiones son:
Los dos grandes defectos de los diabéticos tipo 2 son la resistencia a la insulina y la deficiencia de insulina.
En un estudio longitudinal realizado en Arizona, Estados Unidos, en una cohorte de indios pima, población muy estudiada por su elevada prevalencia de obesidad, se evaluó en cada uno de los casos la sensibilidad a la insulina y el índice de respuesta a la insulina, o la función de las células beta mediante la determinación de la respuesta aguda de insulina a glucosa endovenosa. Se encontró que, en individuos no diabéticos, a medida que disminuye la sensibilidad a la insulina aumenta la respuesta insulínica aguda en forma lineal. Este fenómeno se denomina hiperinsulinemia compensatoria, y se debe a la adaptación de la función de las células beta al fenómeno de resistencia a la insulina(4).
Si los pacientes son tolerantes en forma normal a la glucosa a nivel basal, aunque se vuelvan resistentes durante el seguimiento se mantienen tolerantes en forma normal a la glucosa y, sólo cuando la respuesta insulínica aguda comienza a disminuir, los pacientes se vuelven intolerantes a la glucosa o diabéticos. Esto también es una repetición, mediante información nueva, de conceptos antiguos sobre la insulinorresistencia.
Según datos publicados en San Antonio, Texas, si se hace una prueba de tolerancia a la glucosa oral y se grafica la insulina sobre la concentración de glucosa a los 30 minutos, que es una respuesta relativamente temprana o aguda de la insulina durante una prueba oral de glucosa, y sobre la concentración de glucosa a las dos horas, se puede observar una asociación estadísticamente significativa entre las dos variables.
Esto es interesante, ya que indica que la respuesta de insulina determina una escasa hiperglicemia tardía, lo que se debe a que las células beta tienen una amplia reserva para acomodar un defecto o reducción de la respuesta aguda de insulina. Si se baja hasta cierto nivel la hiperglicemia, se desarrolla otra vez precipitadamente y la característica no lineal de la función indica precisamente que el paciente puede presentar una respuesta de insulina cada vez menor hasta cierto nivel, pasado el cual ocurre un cambio que determina que se vuelva hiperglicémico. Esto indica que la célula beta es un computador muy complejo que se adapta hasta un cierto nivel y cuando se pierde esta adaptación aparece la hiperglicemia.
El hecho de que, cuando disminuye la respuesta aguda de insulina, la glucosa se eleva dos horas después es algo conocido y el único mecanismo fisiológico que puede explicar esto es el efecto de la insulina en la producción de glucosa hepática. La insulina es secretada a la vena porta para que alcance el hígado de forma más rápida y en niveles más altos que los de la circulación sistémica; por lo que el hígado está ubicado en una posición estratégica entre el páncreas y los tejidos periféricos.
La acción de la insulina en el hígado es bloquear la liberación endógena de glucosa. Esto lo hace muy bien y rápido en los no diabéticos, como se demostró en un antiguo experimento en el se hizo un clamp de hiperglicemia tres veces en un no diabético. En el primer estudio se permitió el desarrollo de la respuesta normal endógena de insulina; para esto se usó somatostatina, que bloquea la liberación de insulina exclusivamente en el tiempo en que se inyecta, con el fin de bloquear la respuesta aguda de insulina. En el segundo estudio, la fase temprana de liberación de insulina, que corresponde a la insulina liberada en los diez primeros minutos después de un paso de hiperglicemia, se bloqueó con la somatostatina. El tercer estudio es un control posterior con somatostatina y además con reemplazo de insulina, para tratar de imitar la respuesta fisiológica.
Después de esto se estudió el efecto en la producción hepática de glucosa, que en condiciones fisiológicas se inhibe en forma rápida y potente por acción de la insulina. Si se bloquea la respuesta aguda de insulina, hay muy poco cambio en la producción de glucosa endógena, pero si se libera la respuesta aguda de insulina, entonces la producción de glucosa hepática queda nuevamente suprimida. La liberación de insulina de la fase tardía fue similar en los tres estudios, igual que la producción de glucosa hepática(5).
Estos estudios realizados en seres humanos, lo mismo que los de animales, demuestran que el páncreas libera insulina en respuesta a la glucosa endovenosa, con el objeto de inhibir rápidamente la producción de glucosa hepática y, de esa manera, el organismo maneja la glucosa que ingresa de una manera mucho más eficaz, porque no tiene que manejar dos fuentes distintas de glucosa.
Otro estudio muy interesante, publicado hace 15 años por el grupo australiano de Bruce, tomó a un grupo de pacientes con diabetes mellitus tipo 2, a quienes se administró una comida mixta y luego se les midieron los cambios en los niveles de glicemia e insulinemia durante tres horas (Figura 4). En otras palabras, estudiaron a diabéticos, sin hacer nada, y observaron que la respuesta de glucosa fue alta y tardía, y que la respuesta de insulina fue tardía y menor(6).
Luego tomaron a estos pacientes nuevamente y les reemplazaron la insulina con una infusión endovenosa constante o con una administración tardía, y observaron que el efecto en el perfil de glucosa plasmática fue muy bajo, a pesar de que les dieron más insulina, lo que indica que si se administra insulina suplementaria a diabéticos, ya sea en forma constante, cosa que el páncreas no hace, o en forma tardía, no se corrige la hiperglicemia y sí se produce una hiperinsulinemia, de modo que se obtiene el efecto perjudicial del tratamiento sin lograr su beneficio.
En cambio, si se entrega la misma dosis de insulina en un plazo lo bastante breve después de la comida, de tal manera que se imite la respuesta de insulina plasmática que se ve en los no diabéticos, se observa una mejoría significativa de la hiperglicemia postprandial, aunque no llega a ser normal. Este concepto es importante: si se reemplaza la primera fase de liberación de insulina pueden mejorar las cosas, pero no se puede curar la diabetes.
En un estudio en el que se utilizó insulina regular o insulina lispro, en pacientes levemente diabéticos, sometidos a una comida mixta o a una carga de glucosa oral, y se observaron las respuestas de glucosa e insulina plasmáticas, se comprobó que con lispro, que se absorbe más rápidamente, el resultado del perfil de insulina plasmática se traslada a la izquierda del de la insulina regular(7).
También se midió la producción de glucosa hepática y se observó que con lispro hubo una supresión rápida y completa de esta producción, que fue significativamente mayor que la supresión con insulina regular (Figuras 5 y 6) . En consecuencia, el control de la glicemia fue mejor con lispro que con insulina regular, debido exclusivamente a la cinética de este tipo de insulina. Ésta es otra manera de demostrar que no es tan importante cuánta insulina tiene el cuerpo sino cuánta está disponible para mejorar el control de la glicemia.
Función de las células beta en la diabetes
En el UKPDS (U.K. Prospective Diabetes Study) se reunió a alrededor de 5.000 diabéticos recién diagnosticados, a quienes se hizo una estimación de la función de células beta al inicio del estudio y luego una vez al año, durante seis años. Se observó que la función de las células beta ya estaba reducida al inicio en 50%, en promedio, y que en el transcurso de seis años se redujo a 25%, aproximadamente. Hay que tener presente que algunos de estos pacientes fueron diagnosticados hace 20 años, según criterios que en la actualidad no se usan para diagnosticar la diabetes, por lo tanto en el grupo había una mezcla de hiperglicemias graves, moderadas y normales(8).
Al extrapolar esta información hacia atrás en el tiempo se observa que la función de las células beta estaba intacta 12 años antes del diagnóstico (Figura 7), lo que significa que hay un tiempo en la vida del diabético en que la función de las células beta es normal y luego hay un plazo largo, que antiguamente se conocía como prediabetes, palabra que, lamentablemente, ha sido abandonada, también por causa de los norteamericanos, para describir el tiempo en que se produce una pérdida gradual de esta función, hasta que el problema se manifiesta clínicamente y se hace el diagnóstico de diabetes.
En este momento, el juego ya está medio perdido, porque solamente queda 50% de la función, independientemente de lo que se haga. En el UKPDS se usó dieta, sulfonilureas, metformina e insulina, in crescendo y en combinaciones, para tratar la diabetes, a pesar de lo cual, a los seis años, sólo quedaba 24% de la función. A estos pacientes, en realidad, se les siguió durante 12 años, pero no se graficaron porque el valor se haría negativo. Entonces, las células beta no solamente no estaban produciendo insulina, sino que además sacaban insulina del ambiente.
Frente a esto, es necesario determinar si realmente las células beta están muertas en el paciente diabético, porque, si es así, el estímulo de la célula beta con cualquier droga podría producir más daño que beneficio, al aumentar el estrés en una célula que ya está moribunda. Sin embargo, puede que esto no sea tan así, ya que hay estudios en la literatura que han evaluado la masa de células beta en el páncreas de pacientes a punto de morir, con y sin diabetes.
En el estudio que realizó un grupo de patólogos belgas se describe la información procedente del mayor grupo de pruebas histológicas e histoquímicas hechas en páncreas humanos de individuos diabéticos y no diabéticos. El estudio compara al grupo diabético y al no diabético en los mismos términos de IMC, para lo cual los grupos se dividieron en obesos y delgados para evitar confusiones, ya que la obesidad aumenta la masa de células beta porque ocurre una hipertrofia celular en respuesta a la obesidad y dificulta la comparación (Figura 8). Encontraron que, salvo muy pocas excepciones de pacientes que usaban insulina cuando estaban vivos, la masa de células beta de los pacientes con diabetes tipo 2 estaba preservada y no tenía diferencia estadísticamente significativa con la masa de células beta de los no diabéticos, ya fueran delgados u obesos(9).
Se ha hablado mucho acerca del procesamiento de la insulina y los niveles de proinsulina, que están altos en los diabéticos y son predictores de enfermedades cardiovasculares, según muchos estudios. Pues bien, en el páncreas de los diabéticos tipo 2 obesos, hay alguna evidencia de un déficit de procesamiento, de una falta de capacidad para procesar la prohormona hacia la hormona natural, pero estos páncreas aún se tiñen para insulina, de modo que aún pueden secretar o sintetizar la prohormona.
Otro tema es el de los amiloides. Se han producido grandes discusiones con respecto a la infiltración amiloídea en los islotes de Langerhans, en diabéticos, como un mecanismo de destrucción de las células beta en los diabéticos tipo 2. Hay muchos modelos de animales, por ejemplo ratones, sobre expuestos selectivamente a amiloides en las células beta de los islotes de Langerhans, que han demostrado que a medida que el amiloide se deposita, la célula beta muere.
Sin embargo, parece que no ocurre lo mismo en los seres humanos, pues, al medir la cantidad de amiloide en pacientes control y en pacientes con diabetes, se ve que el porcentaje de controles con amiloide es un tercio, lo que significa que un tercio de nosotros tendrá algún grado de infiltración amiloídea en las células beta, a pesar de no tener diabetes. Por su parte, el porcentaje de diabéticos con amiloide no es mucho más alto y, lo que es más importante, los islotes teñidos positivos para amiloide todavía se tiñen positivos para insulina, lo que significa que la presencia de amiloide no es una anormalidad que indique forzosamente que la célula beta esté muerta.
El concepto que está surgiendo, y que probablemente se hará más fuerte en el futuro, es que la masa de células beta está bastante preservada en los diabéticos tipo 2 y que todas las alteraciones histoquímicas que se han descrito en modelos de animales, como la infiltración amiloídea, la destrucción de islotes, etc., no tiene un significado patológico real en seres humanos.
Estudio de función de las células beta
Si se acepta que la masa de células beta está preservada en los diabéticos, aún queda el problema funcional: están, pero no funcionan. Para determinar el grado de sensibilidad a la insulina o el nivel de insulinorresistencia, se pueden hacer varias pruebas. Por ejemplo, se puede hacer un clamp, que dará un número que se ha validado en cientos de pacientes y que indicará el grado de sensibilidad a la insulina dentro de una escala; pero no hay algo similar para la secreción de insulina. No se puede recomendar una prueba específica para medir el grado de función de las células beta, porque se puede hacer un test con infusión de glucosa endovenosa (IVGTT), test de tolerancia a la glucosa oral (OGTT) o pruebas de comida, y obtener resultados diferentes.
En el clásico estudio de Ward, se administró glucosa endovenosa en bolos y se observó la famosa fase temprana de respuesta de insulina en individuos no diabéticos, que estaba completamente perdida en los pacientes diabéticos tipo 2 (Figura 9). Con este criterio, grupos de personas que se estudiaron hace 20 años son deficientes en insulina, sólo porque no respondieron a bolos endovenosos(10).
En otro estudio realizado en 33 pacientes típicos con diabetes tipo 2: se les administró glucosa endovenosa a tiempo cero, otra vez el clásico IVGTT con 20 gramos, como bolo, y se midió la concentración de péptido C para reconstruir los rangos de secreción de insulina; en vez de la concentración de insulina plasmática, se ven los rangos de secreción de insulina en picomoles de hormona por minuto. En los diabéticos se ve una respuesta de primera fase de insulina muy lenta y luego una fase inhibitoria (Figura 10).
Este fenómeno ya se había comunicado antes, así que no causa sorpresa ver que la glucosa endovenosa produce inhibición de la producción de insulina, en ayunas, en pacientes con diabetes. Si se usa este criterio como guía, habría que decir que estos 33 pacientes con diabetes tipo 2 son completamente incompetentes en términos de respuesta de células beta a la glucosa, pero si a las mismas personas se les administra glucosa oral, ellas responden muy bien y producen mucha insulina. La diferencia entre los dos resultados está en la forma de administrar la glucosa, no en la función de las células beta.
Prueba de tolerancia a la glucosa oral
Todos los datos clásicos sobre respuesta a insulina frente a la administración de glucosa oral, en pacientes normales y diabéticos, delgados y obesos, demuestran una respuesta de insulina insuficiente. Con un clamp de hiperglicemia se pueden elevar la concentraciones de glucosa plasmática a 175 mg/dl, en personas con tolerancia normal a la glucosa y con diabetes tipo 2 o tolerancia disminuida a glucosa, y se obtiene una respuesta reducida en IGT y ninguna respuesta en diabéticos(11).
El problema con todas estas pruebas es que la secreción absoluta de insulina no es un buen índice de función de las células beta, si no se compara con los niveles de glucosa; es como comparar peras con manzanas. Para comparar secreción de insulina se debe tomar en cuenta la concentración de glucosa plasmática; la comparación directa de la secreción absoluta de insulina, entre grupos con diferentes niveles de glucosa, no es lo apropiado.
La mejor manera de hacerlo es volver a los experimentos con comidas mixtas y desarrollar un modelo que relacione la concentración de glucosa plasmática con la respuesta de insulina plasmática. Esto no es nada nuevo, porque lo hizo Bagdade, en páncreas aislado perfundido, en los años 60(12). Lo importante es recordar que sólo se puede comparar la secreción de insulina si se toman en cuenta los niveles de glucosa plasmáticos concomitantes.
Respuesta de las células beta a la glucosa
En nuestro estudio se analizaron los rangos de secreción de insulina, reconstruidos a partir de la concentración de péptidos C, durante 15 horas de vida regular con tres comidas y vida libre, en sujetos no diabéticos y en un grupo grande de 59 pacientes con diabetes tipo 2 (Figura 11).
Con estos datos se llegó al concepto de que, en términos absolutos, los pacientes no secretan poca insulina sino que, al contrario, secretan más (Figura 12).
Analizando cómo una célula beta en cultivo responde a la glucosa, se pueden apreciar las características principales del computador de una célula beta. Esta célula debe ser capaz de percibir la concentración de glucosa, de modo que, mientras más alta sea ésta, produzca mayor secreción de insulina. Esta función se llama función de dosis/respuesta y es clásica (Figura 13).
La célula beta también responde a los cambios de la glucosa mediante una memoria que le permite recordar cuál era el nivel de glucosa a un tiempo menos t y sobre esa base responde a tiempo t. Esto tampoco es nuevo y se puede demostrar rápidamente en un páncreas perfundido aislado, tanto en ratas como en seres humanos. El resultado recibe el nombre de elemento derivativo o elemento dinámico de la liberación de insulina en respuesta a glucosa.
Por último, existe el concepto de potenciación, que se refiere simplemente al hecho de que, después de la primera comida, la respuesta a la comida siguiente será mejor, debido a la memoria de largo plazo del nivel de glucosa, que tiene la célula beta, y a la potenciación.
La potenciación ayuda a diferenciar entre la infusión de glucosa y la ingesta de glucosa. Este concepto tampoco es nuevo; cuando la glucosa ingerida toma contacto con el epitelio intestinal y éste la absorbe, estimula la liberación de numerosas hormonas, como GIP, colecistoquinina, enzimas pancreáticas, gastrina, insulina y GLP-1 (Glucagon-like peptide), todas las cuales potencian la liberación de insulina inducida por la glucosa. En forma aislada no hacen gran cosa, pero, en conjunto con la hiperglicemia, estimulan la liberación de insulina.
Por lo tanto, existen a lo menos tres fenómenos básicos que se deben considerar cuando se relaciona la glucosa plasmática con la secreción de insulina: el nivel de glucosa, el cambio en un plazo y la potenciación.
Al analizar el rango de secreción de insulina y dividir a los pacientes según el tipo de hiperglicemia (hiperglicemia en ayunas, hiperglicemia leve e hiperglicemia grave), se puede ver que ni la respuesta basal de insulina ni la cantidad total de insulina producida durante 15 horas, en respuesta a tres comidas mixtas, está reducida en los diabéticos, en comparación con los no diabéticos (Figura 14).
Si luego se grafican las curvas de dosis/respuesta obtenidas con el modelo matemático, se puede ver que frente a cualquier nivel de glucosa, la cantidad de insulina secretada es mucho menor en los diabéticos que en los no diabéticos. Otra manera de decirlo es que la función de dosis/respuesta se traslada hacia la derecha en los diabéticos, en relación a los no diabéticos, y la pendiente es menos marcada (Figura 15).
Por otra parte, se puede ver que cuanto más grave es la hiperglicemia, más plana es la curva de dosis/respuesta (Figura 16). Por lo tanto, no es lo mismo que el diabético sea un hiperglicémico leve por la mañana, cuando se despierta a desayunar, ya que responderá mejor a la glucosa, o que sea un hiperglicémico grave, que responderá lentamente.
Si luego se grafica el componente derivativo, es decir, la capacidad de la célula beta para responder rápidamente, se ve que en los diabéticos esta capacidad está deprimida o perdida, a pesar de la gravedad de la hiperglicemia (Figura 17), lo que sugiere que la capacidad de la célula beta para responder agudamente, con un aumento rápido de la secreción de insulina, se pierde pronto en la evolución de la enfermedad, de modo que cuando se encuentra una hiperglicemia importante en ayunas, es decir, diabetes manifiesta, esa función ya está disminuida.
El fenómeno de potenciación actúa de tal forma que, en los pacientes control, la primera comida, el desayuno, que no es tan grande, potencia la respuesta posterior de insulina ante las demás comidas. Este fenómeno de potenciación se traslada completamente hacia la derecha y disminuye en los diabéticos (Figura 18).
Hoy se sabe que la liberación del GLP-1 (péptido glucoquinina-símil tipo uno) en respuesta a glucosa oral es insuficiente en los diabéticos y que, por lo tanto, el efecto de potenciación de esta hormona intestinal en la secreción de insulina inducida por glucosa está disminuida en la diabetes. Por este motivo, la industria está desarrollando análogos de GLP-1 o compuestos que puedan disminuir la velocidad de degradación de GLP-1 para tratar la diabetes.
La potenciación no sólo tiene interés fisiológico, sino que también es un objetivo para el tratamiento. El ideal es tener una droga que logre restablecer la dinamia de la secreción de insulina, es decir, que estimule rápidamente la secreción de insulina por parte de las células beta, después de la comida, pero que detenga la secreción relativamente pronto, de manera que inicialmente se libere más insulina y luego, menos.
Básicamente, el ideal es tratar los dos problemas de la diabetes, la hipoinsulinemia que alterna con la hiperinsulinemia, y revertirlos. Con esto tendría que mejorar el control de la glucosa, incluso se puede tomar el resultado como prueba del mecanismo de acción de la droga, pero aunque mejore el control la glucosa no se normalizaría totalmente, ya que este problema no es el único.
A un grupo de pacientes definidos como diabéticos, que tenían una hiperglicemia leve en ayunas y que se asignaron al azar a cuatro dosis distintas de la droga, se les estudió a nivel basal y seis meses más tarde. Al analizar la respuesta, cuatro horas después de una cena mixta, en el grupo placebo se observó que después de seis meses la respuesta a la glucosa fue peor que al inicio, debido al fenómeno conocido como “agotamiento del placebo” (figura 19).
Se presenta la información preliminar en diez casos del grupo que recibió 120 mg de nateglanide, a tiempo cero, en la comida (figura 20). El péptido C estuvo levemente más alto durante la primera media hora, igual que la insulina, pero el perfil de glucosa plasmática, a los seis meses, fue mucho menor que el nivel basal y la respuesta fue mayor que con placebo.
Esto no indica forzosamente que la función de las células beta está alterada, puesto que la respuesta, en términos de péptido C o insulina, es pequeña frente a niveles bajos de glucosa y, por lo tanto, las diferencias son pequeñas, de modo que se necesita un modelo para determinar si hubo o no algún efecto sobre la función de las células beta (Figura 21). Al analizar la información con este modelo, se observó que aquellas funciones de respuesta, que eran planas antes del tratamiento, a los seis meses se trasladaron significativamente hacia arriba y que la respuesta a una dosis completa fue más alta, lo que indica que, frente a cada concentración de glucosa plasmática, la secreción de insulina fue más alta con la droga que sin ella, o sea, la nateglinida aumentó la respuesta a una determinada dosis de glucosa.
La potenciación no cambió (Figura 22), pero el componente dinámico, la capacidad de responder rápidamente, fue mejorando de manera significativa, hasta duplicarse a las 24 semanas de tratamiento (Figura 23).
En este estudio se concluyó que la nateglinida tiene un efecto específico a nivel de la fase temprana de la liberación de insulina, en la capacidad de captar el cambio de glucosa y responder rápidamente.
Esto no es nuevo, porque la tolbutamida también tiene ese efecto y este fármaco pertenece a la primera generación de sulfonilureas, las que actuaban tan rápidamente que se tenían que administrar tres veces al día. Después se desarrolló el concepto de que las drogas se debían administrar sólo una vez al día, para que los pacientes no perdieran la adhesión al tratamiento, y entonces se desarrollaron las sulfonilureas de segunda generación, que se administraban una vez al día y estimulaban la liberación de insulina inducida por glucosa, durante las 24 horas, y producían hipoglicemias entre las comidas.
Por lo tanto, estamos volviendo al pasado, porque ya había compuestos capaces de estimular específicamente la célula beta cuando fuera necesario.
La otra información que se obtiene de este estudio es que, con la intervención, la curva completa de dosis/respuesta se traslada hacia arriba, lo que significa que la célula beta puede funcionar de distintas maneras, pero todas están interrelacionadas y, si se logra mejorar una, también lo harán las otras.
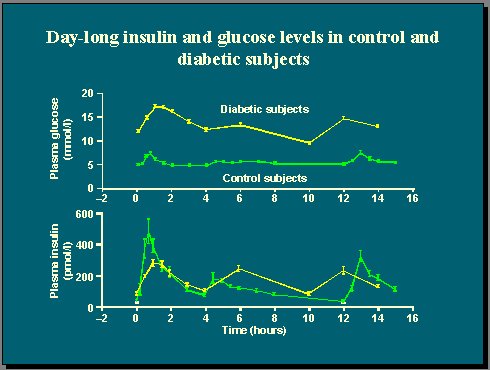 Figura 1.
Figura 1.
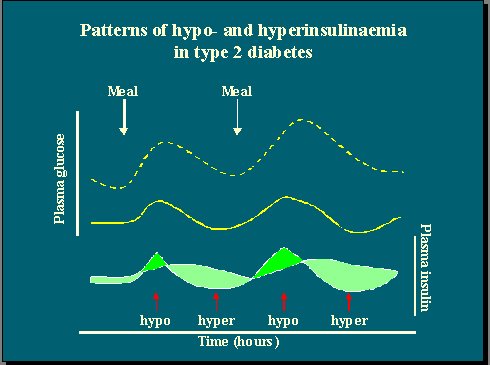 Figura 2.
Figura 2.
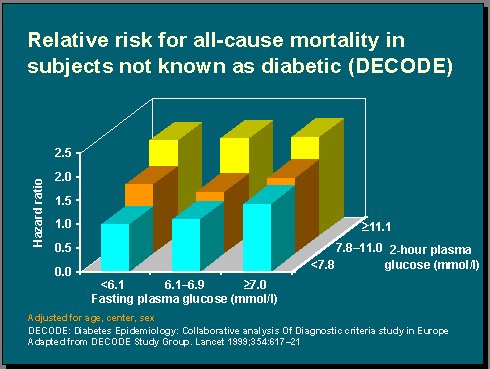 Figura 3.
Figura 3.
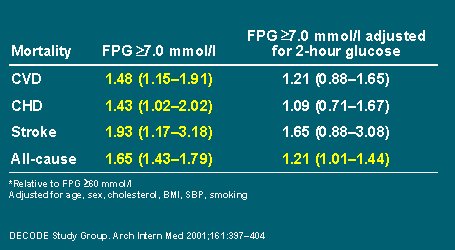 Tabla I. Riesgo relativo* de mortalidad en pacientes diabéticos asintomáticos según FPG (ADA).
Tabla I. Riesgo relativo* de mortalidad en pacientes diabéticos asintomáticos según FPG (ADA).
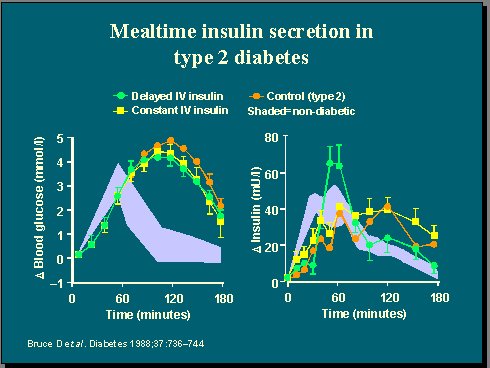 Figura 4.
Figura 4.
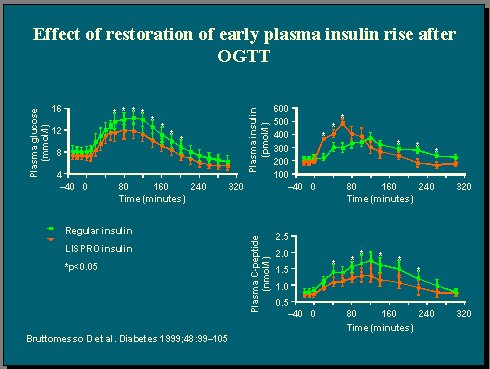 Figura 5.
Figura 5.
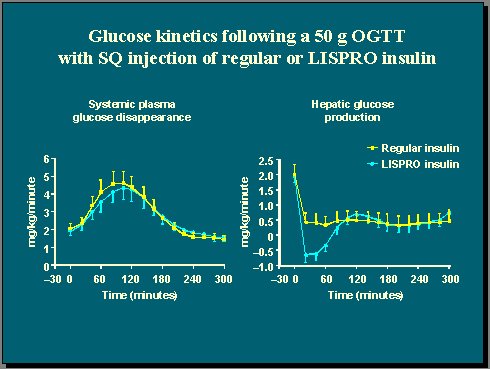 Figura 6.
Figura 6.
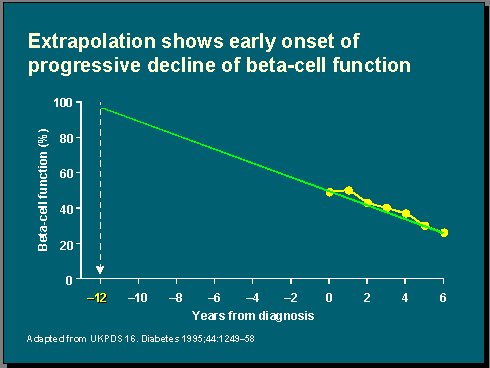 Figura 7.
Figura 7.
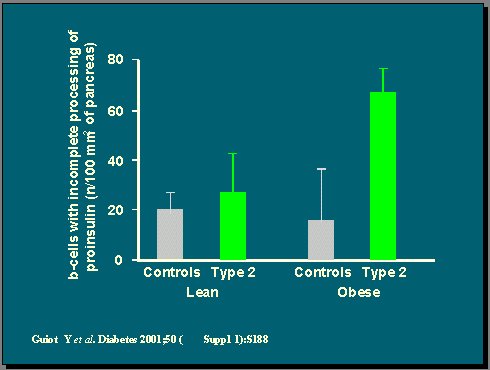 Figura 8.
Figura 8.
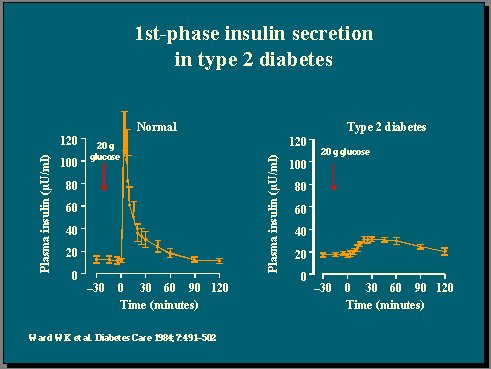 Figura 9.
Figura 9.
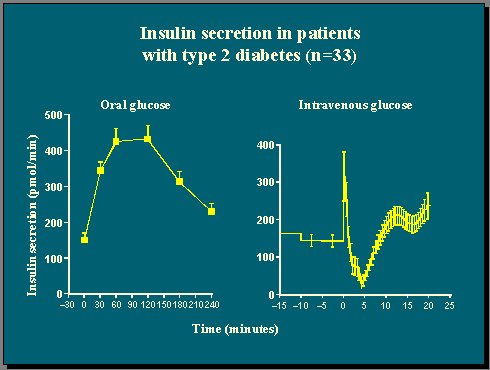 Figura 10.
Figura 10.
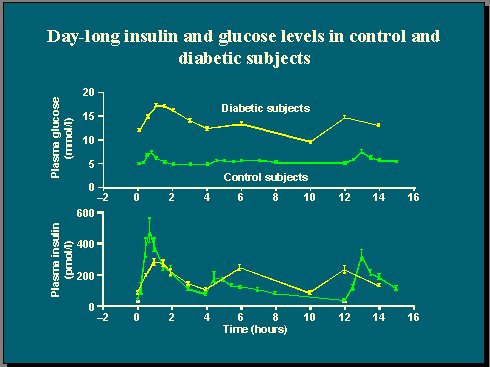 Figura 11.
Figura 11.
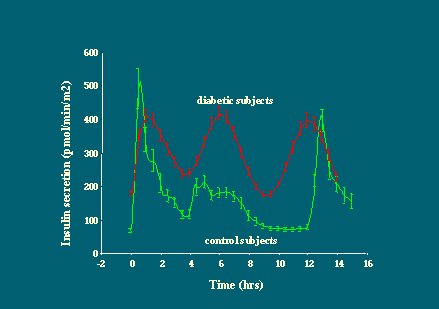 Figura 12.
Figura 12.
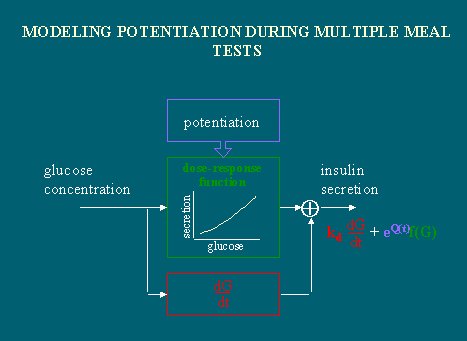 Figura 13.
Figura 13.
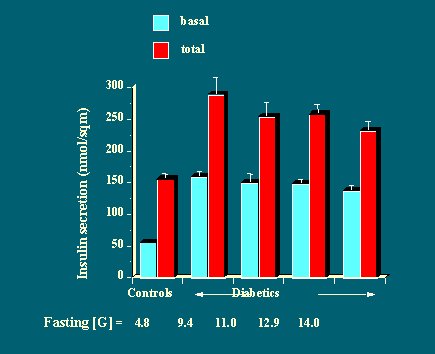 Figura 14.
Figura 14.
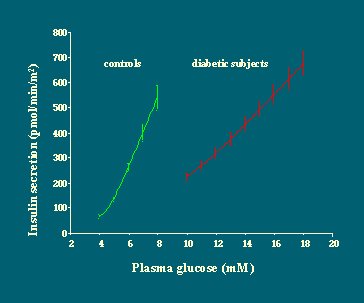 Figura 15.
Figura 15.
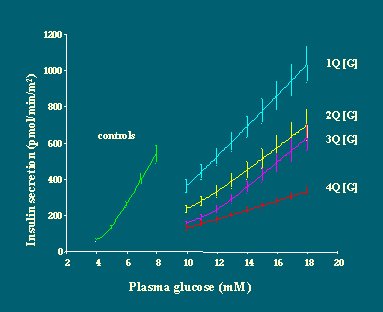 Figura 16.
Figura 16.
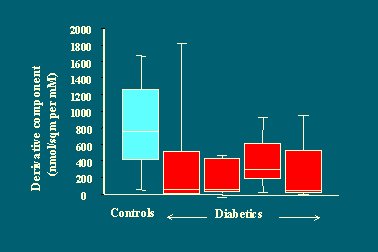 Figura 17.
Figura 17.
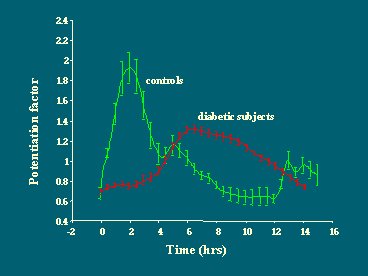 Figura 18.
Figura 18.
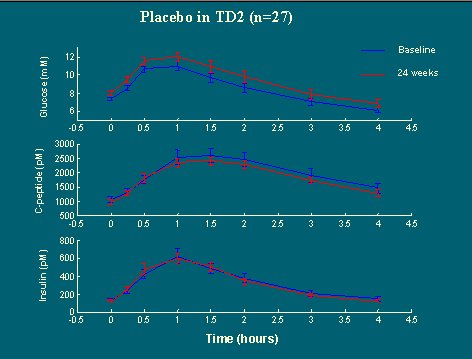 Figura 19.
Figura 19.
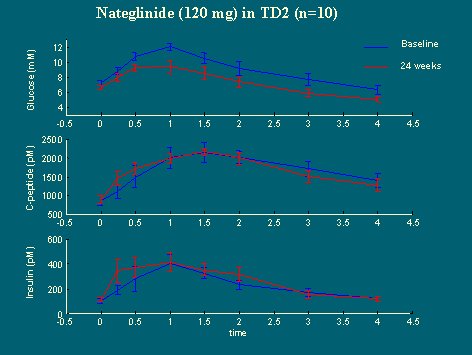 Figura 20.
Figura 20.
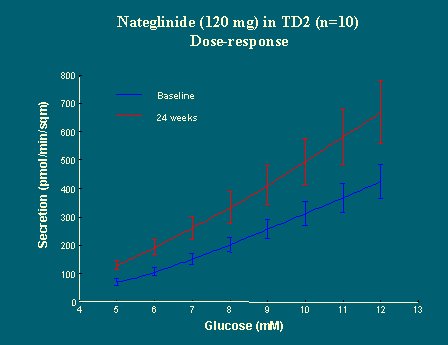 Figura 21.
Figura 21.
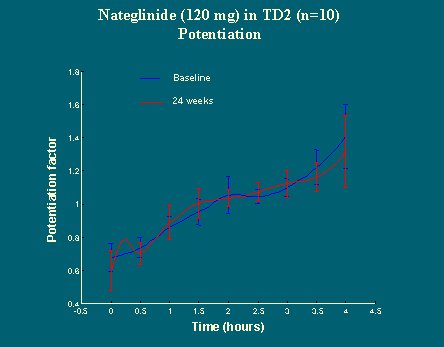 Figura 22.
Figura 22.
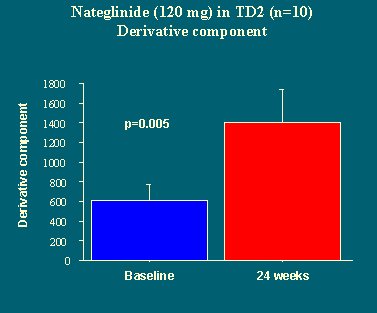 Figura 23.
Figura 23.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Este texto completo es la transcripción editada y revisada de la conferencia dictada en el marco del Simposio Hiperglicemia Postprandial, Factor Emergente de Riesgo Cardiovascular en la Diabetes Tipo 2, realizado el 11 de abril de 2002. El evento fue organizado por Novartis.
Expositor:
Ele Ferrannini[1]
Citación: Ferrannini E. Postprandial hyperglycemia, an emerging cardiovascular risk factor in Type 2 (2). Medwave 2002 Jun;2(5):e603 doi: 10.5867/medwave.2002.05.603
Fecha de publicación: 1/6/2002
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Glucose tolerance and mortality: comparison of WHO and American Diabetes Association diagnostic criteria. The DECODE study group. Lancet 1999 Aug 21;354(9179):617-21. | CrossRef |DECODE Study Group, Arch Intern Med 2001;161:397-404. Hanefeld M, Fischer S, Julius U, Schulze J, Schwanebeck U, Schmechel H, et al. Risk factors for myocardial infarction and death in newly detected NIDDM: the Diabetes Intervention Study, 11-year follow-up. DiabetologĂa 1996 Dec;39(12):1577-83. | CrossRef | PubMed |Lillioja S, Mott DM, Spraul M, Ferraro R, Foley JE, Ravussin E, et al Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians”. N Eng J Med 1993 Dec 30;329(27):1988-92.
| CrossRef | PubMed |Luzi L, DeFronzo RA. Effect of loss of first-phase insulin secretion on hepatic glucose production and tissue glucose disposal in humans. Am J Physiol 1989;257:E241-246. Bruce DG, Chisholm DJ, Storlien LH, Kraegen EW. “Physiological importance of deficiency in early prandial insulin secretion in non-insulin-dependent diabetes”, Diabetes 1988 Jun;37(6):736-44.
| CrossRef | PubMed |Bruttomesso D, Pianta A, Mari A, Valerio A, Marescotti MC, Avogaro A, et al. Restoration of early rise in plasma insulin levels improves the glucose tolerance of type 2 diabetic patients. Diabetes 1999; 48:99-105 U.K. prospective diabetes study 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group. Diabetes 1995 Nov; 44(11):1249-58. | CrossRef | PubMed |Guiot Y, Sempoux C, Moulin P, Rahier J. No decrease of the beta-cell mass in type 2 diabetic patients. Diabetes 2001 Feb;50 Suppl 1:S188. | CrossRef |Ward WK, Beard JC, Halter JB, Pfeifer MA, Porte D Jr. Pathophysiology of insulin secretion in non-insulin-dependent diabetes mellitus. Diabetes Care 1984;7:491-502. 






 Estudios originales
Estudios originales