Resumen
Este texto completo es la transcripci¾n editada y revisada de la conferencia dictada en el marco del V Congreso de Obstetricia, GinecologĒa Infantil y Adolescencia, realizado en Santiago entre los dĒas 31 de agosto al 2 de septiembre de 2006. El evento fue organizado por la Sociedad Chilena de Obstetricia, GinecologĒa Infantil y Adolescencia.
Presidente: Dra. Pamela Oyarz·n.
Introducci¾n
Hasta agosto de 2005, habĒa en
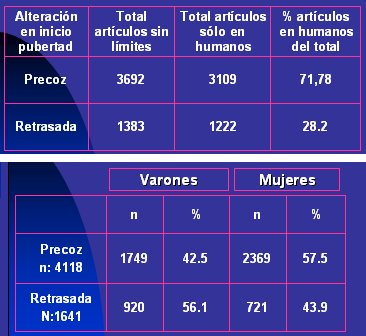
Pubmed alrededor de 3.700 artĒculos relacionados con alteraciones en el inicio de la pubertad, entre ellos algunos que comunicaban experiencias en animales. 3.100 artĒculos se referĒan a estudios en seres humanos y de ellos, 72 trataban sobre pubertad precoz y 28%, sobre pubertad retrasada (Tabla I, arriba). En la b·squeda por sexo, como la pubertad retrasada afecta mayoritariamente al sexo masculino, el porcentaje de artĒculos sobre el problema en varones fue de 56,1% (Tabla I, abajo).
Tabla I. Resultado de b·squeda sobre alteraciones de la pubertad (Pubmed, agosto 2005)
Pubertad retrasada
La
pubertad retrasada es la ausencia de caracteres sexuales secundarios, a 2 desviaciones estßndar de la edad promedio del inicio de la pubertad normal en la poblaci¾n y sexo a los que pertenece el individuo. En tķrminos prßcticos y cronol¾gicos, para los varones es a los 14 a±os, cuando el volumen testicular es menor de 4 ml y para las mujeres es a los 13 a±os, cuando a·n no hay presencia de telarquia. Una situaci¾n especial es la
pubertad detenida, que corresponde a un grupo de pacientes que pueden tener anormalidades parciales o transitorias; inician la pubertad a una edad normal, pero transcurren mßs de cinco a±os entre el primer signo puberal y el desarrollo gonadal completo en hombres o la aparici¾n de menarquia, en mujeres.
La pubertad retrasada es un cuadro clĒnico frecuente que ocurre en 3% de la poblaci¾n, pero hay pocos estudios, y los que hay no son homogķneos, en el anßlisis de las diferentes causas de pubertad retrasada; tambiķn es mßs frecuente en varones, pero en ambos sexos la variedad mßs frecuente es la pubertad retrasada de tipo simple, que por lo general es de origen familiar o idiomßtica y se debe a retraso constitucional del crecimiento y la pubertad. 60% de los casos se da en varones y 30%, en mujeres.
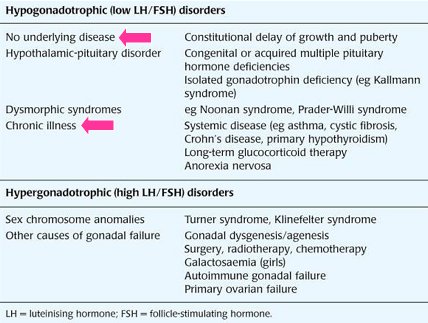
Las causas mßs frecuentes de pubertad retrasada son: las enfermedades cr¾nicas, como asma, enfermedad celĒaca, enfermedad de Crohn, hipotiroidismo primario, terapia prolongada con glucocorticoides, anorexia nerviosa; y los cuadros fisiol¾gicos que no representan enfermedad, como el retraso constitucional del crecimiento y la pubertad. Otras patologĒas constituyen causas de pubertad retrasada poco frecuentes, pero es un gran logro conocerlas en detalle, pues su estudio molecular ha permitido entender el complejo fen¾meno de la pubertad; entre ellas se pueden mencionar: los trastornos del eje hip¾fisis-hipotßlamo, como el sĒndrome de Kallmann y las deficiencias congķnitas o adquiridas de hormonas hipofisiarias; sĒndromes dism¾rficos, como los sĒndromes de Noonan y de Prader-Willi; anomalĒas en los cromosomas sexuales, como los sĒndromes de Turner y Klinefelter); y otras causas de insuficiencia gonadal, como agķnesis-disgķnesis gonadal, cirugĒa, radioterapia, quimioterapia, insuficiencia ovßrica primaria, insuficiencia gonadal autoinmune y galactosemia, en las ni±as. En la Tabla II se resumen las causas de pubertad retrasada y detenida.
Tabla II. Principales causas de pubertad retrasada y detenida
Genķtica de la pubertad retrasada
El genotipo en las alteraciones del eje HHG se resume de la siguiente manera:
- Hipotßlamo: GnRH/migraci¾n; defectos en la sĒntesis y liberaci¾n de GnRH; alteraciones de leptina y su receptor; alteraciones de factores de transcripci¾n.
- Hip¾fisis: Mutaciones en el receptor de GnRH; alteraciones en el desarrollo; alteraciones en producci¾n hormonal
- G¾nadas: Alteraciones en las subunidades de FSH y LH; mutaciones en los receptores FSH y LH; alteraciones en las se±ales intracelulares; alteraciones en la funci¾n y diferenciaci¾n.
Para entender las causas de la pubertad retrasada se debe conocer las bases gķnicas, especialmente los siguientes genes:
- De la migraci¾n de las neuronas que producen hormona liberadora de gonadotrofinas (GnRH): gen KAL
- De la acci¾n del GnRH: gen del receptor de GnRH
- De sĒntesis de gonadotrofinas: genes de subunidades de gonadotrofina
- De acci¾n de las gonadotrofinas: genes del receptor de gonadotrofinas.
En cuanto a los defectos de la migraci¾n de neuronas productoras de GnRH, el gen KAL se localiza en la regi¾n pseudoautos¾mica del cromosoma X (Xp22.3); codifica para Anosmin-1 y para la glicoproteĒna de matriz extracelular, encargadas del crecimiento y migraci¾n de neuronas olfatorias y productoras de GnRH; este gen origina el fenotipo del sĒndrome de Kallmann, un hipogonadismo hipogonadotr¾fico que se caracteriza por anosmia o hiposmia, fisura labiopalatina, sordera congķnita, convulsiones cerebelosas, cuarto metacarpiano corto y agenesia renal, en 50% de los casos. El gen KAL se ubica en el cromosoma X y su expresi¾n es fundamental los siguientes sitios (lo que explica las manifestaciones clĒnicas): cerebelo (sincinecia), n·cleo ¾ptico (anomalĒas visuales), mesķnquima facial (defectos de la lĒnea media facial) y meso-metanefro (agenesia renal). En cuanto a defectos en la sĒntesis y liberaci¾n de GnRH, no se han descrito alteraciones que provoquen alguna enfermedad en el ser humano; solamente se ha descrito mutaciones en el gen Gn RH con hipogonadismo hipogonadotr¾fico hereditario recesivo en animales de experimentaci¾n, especĒficamente en ratas.
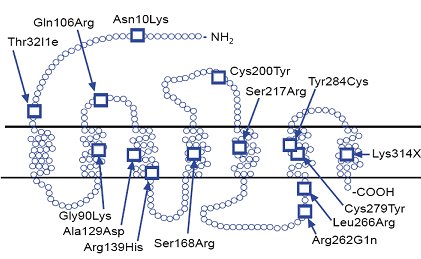
La Fig. 1 es un esquema del gen del receptor de GnRH humano, en el cual se han descrito 14 mutaciones de tipo inactivante que pudieran condicionar casos de pubertad retrasada (1). Las mutaciones del gen del receptor de GnRH se han encontrado en 20% de los hipogonadismos hipogonadotr¾ficos; la mayorĒa de estas mutaciones son heterocigotas y reducen, tanto la uni¾n del GnRH con su receptor, como la activaci¾n de se±ales intracelulares, lo que produce fenotipos muy variables: la pķrdida completa de la funci¾n se ve en la mutaci¾n Ala 1290 Asp / Ser 168 Arg, que da como manifestaciones fenotĒpicas micropene, criptorquidia, ausencia de desarrollo puberal y resistencia al tratamiento con GnRH pulsßtil; otras mutaciones pueden dar compromiso leve de la funci¾n, con fenotipo de desarrollo puberal incompleto, gonadotrofinas basales detectables y modesta respuesta a GnRH pulsßtil.
Figura 1. Mutaciones en el gen del receptor del GnRH humano. Todas inactivantes (1)
En relaci¾n a mutaciones y polimorfimos en los genes de las subunidades alfa y beta de las gonadotrofinas, se sabe que la subunidad alfa es com·n para la hormona luteinizante (LH), la hormona folĒculo estimulante (FSH), la gonadotrofina cori¾nica (hCG) y la hormona estimulante del tiroides (TSH), de modo que una mutaci¾n en esta cadena tendrĒa un resultado ominoso para el paciente. Las mutaciones de las subunidades beta para LH y FSH son las que se acompa±an de pubertades retrasadas. Existe una tipificaci¾n en cuanto a los nucle¾tidos alterados y el intercambio de aminoßcidos; se sabe que la mutaci¾n de la subunidad beta de LH puede provocar falta de inicio espontßneo de la pubertad y que cuando la mutaci¾n se ubica en el ex¾n 2, se presenta un retraso en el inicio de la pubertad. Para la subunidad beta de FSH, algunas mutaciones en el ex¾n 3 tambiķn pueden causar hipogonadismo masculino, con azoospermia e infertilidad y, en la mujer, amenorrea primaria e infertilidad (1).
La mutaci¾n del gen de la subunidad beta de LH se ha descrito solamente en un var¾n, que tenĒa genitales masculinos normales, pubertad retrasada, niveles de LH sķrica elevada y FSH sķrica normal, la cual aument¾ con la edad; ademßs, este paciente tenĒa espermatogķnesis disminuida y ausencia de cķlulas de Leydig. Cuando se le realiz¾ una prueba de estimulaci¾n con gonadotrofina cori¾nica se estimul¾ la sĒntesis de testosterona y la espematogķnesis, pero no se logr¾ conseguir fertilidad con esta metodologĒa terapķutica.
Se ha descrito a cinco pacientes con mutaciones de la subunidad beta de FSH: tres mujeres adultas y dos varones, en quienes este gen se localiz¾ en el brazo corto, fracci¾n 13 del cromosoma 11 (11p13). En las mujeres, esta mutaci¾n produjo amenorrea primaria, infertilidad, ausencia de tejido mamario, pubertad retrasada con LH aumentada y FSH disminuida y presencia de glßndulas suprarrenales normales. En los varones se present¾ con pubertad normal, testĒculos peque±os, azoospermia, hipogonadismo secundario a dķficit aislado de FSH, aumento de la LH sķrica y disminuci¾n de la testosterona. Los casos se±alados son situaciones infrecuentes, pero han permitido conocer c¾mo funciona esta cadena de genes en el desarrollo de la pubertad normal.
Las alteraciones de los receptores de gonadotrofinas a nivel gonadal se deben a mutaciones que se localizan en el cromosoma 2. Inicialmente se describieron mutaciones inactivantes en seis familias: en las mujeres habĒa insuficiencia ovßrica primaria, anovulaci¾n, amenorrea primaria o secundaria y presencia de folĒculos primordiales en el ovario; en los hombres las caracterĒsticas sexuales secundarias eran normales, habĒa grados variables de insuficiencia en la espermatogķnesis y testĒculos peque±os, aunque ninguno de los pacientes era azoospķrmico, la FSH sķrica estaba moderadamente aumentada y la LH sķrica estaba normal o moderadamente aumentada.
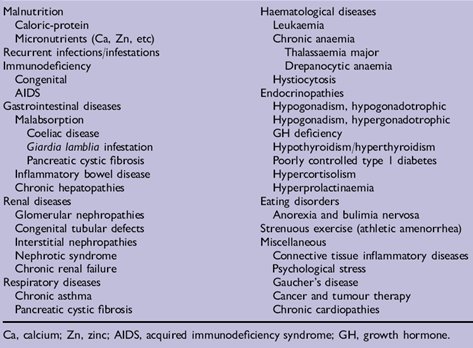
La causa mßs frecuente de pubertad retrasada, ademßs de la constitucional, son las enfermedades cr¾nicas, que pueden determinar que la pubertad se inicie en forma tardĒa. Existe una larga lista de este tipo de enfermedades (Tabla III).
Tabla III. Enfermedades cr¾nicas causantes de pubertad retrasada
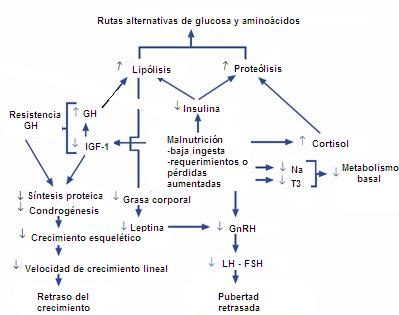
La malnutrici¾n, probablemente por un mecanismo de conservaci¾n, afecta la producci¾n de insulina, del factor de crecimiento insulinosĒmil (IGF-1) y de leptina y disminuye los niveles de hormonas tiroideas, especĒficamente T3 (2). Con mßs detalle, la disminuci¾n de la insulina aumenta la lip¾lisis y la proteolisis, probablemente porque el organismo busca una forma de compensaci¾n de fuente energķtica. La disminuci¾n de la sĒntesis de IGF-1 a nivel hepßtico se compensa con el aumento de la hormona de crecimiento, pero, como hay una disminuci¾n de la sĒntesis de receptores, esto determina un cuadro de resistencia a la hormona de crecimiento y retraso del crecimiento estatural. Por otro lado, si disminuye el tejido graso hay menos leptina, lo que ocasiona retraso de la pubertad. La T3 disminuida se traduce en disminuci¾n del metabolismo basal, de modo que este paciente estķ en situaci¾n de conservaci¾n para un caso de estrķs, en cuanto a nutrici¾n (Fig. 2).
Figura 2. Malnutrici¾n y pubertad retrasada: fisiopatologĒa
Estudio y tratamiento
En el estudio de una pubertad retrasada o detenida, el estudio mĒnimo incluye la determinaci¾n en sangre de: hormonas tiroideas, LH, FSH, testosterona o estradiol cuando corresponda, prolactina y exßmenes generales para descartar una patologĒa cr¾nica, como hemograma-VHS, PCR, funci¾n renal, electrolitos plasmßticos y perfil hepßtico. En las mujeres se debe plantear la posibilidad de hacer un cariograma; en los hombres, ķste se realiza seg·n el fenotipo; en las ni±as se debe hacer, ademßs, una ecotomografĒa uterina y ovßrica.
Una vez que se han descartado las patologĒas mencionadas y se confirma que se trata de un retraso constitucional del crecimiento y la pubertad, se debe decidir si corresponde o no iniciar tratamiento. Por lo general se trata de un escolar var¾n, que estß muy preocupado porque todos sus compa±eros estßn creciendo rßpidamente y ķl a·n no ha dado el estir¾n puberal, ni ha cambiado la voz, ni tiene barba; con frecuencia se le han hecho varios tratamientos para el retraso puberal, con la idea de estimular un hipotßlamo probablemente mßs lento y provocar el inicio de los cambios hormonales propios de la pubertad (3). En las ni±as estß situaci¾n es muy poco frecuente y los endocrin¾logos infantiles la ven poco.
Algunas publicaciones se±alan que algunos ni±os con pubertad retrasada tendrĒan compromiso de su estatura final, porque ōno tienen tiempo suficienteö para alcanzar la estatura que les corresponde por su genķtica y se dice que esta pķrdida, al final del crecimiento, serĒa de alrededor de 2 a 2,5 cms. Por lo tanto, cuando se habla de prescribir el uso de testosterona a un ni±o, sabiendo que es un esteroide sexual, que puede influir en un cierre anticipado de los cartĒlagos de crecimiento, es importante tener claro si es o no necesario su uso y si puede o no ser perjudicial. En un trabajo publicado en 2003 en
Journal of Clinical Endocrinology, se tom¾ a 64 ni±os, separados en un grupo de 23 ni±os control y un grupo de 41 ni±os tratados con enantato de testosterona, en dosis de 125 mg intramuscular, durante tres meses. La talla y la edad de ambos grupos fue similar al inicio del tratamiento; las desviaciones estßndar por debajo de la media tambiķn fueron semejantes y al tķrmino del crecimiento la talla de los ni±os fue muy similar, por lo que estos pacientes no se perjudicaron con un tratamiento que les permiti¾ iniciar la pubertad en forma un poco mßs anticipada que lo que hubiera ocurrido de manera espontßnea (4).
Cuando la pubertad retrasada se debe a una causa que impide la producci¾n de esteroides sexuales, es necesario realizar la inducci¾n de la pubertad, que en la actualidad ha mejorado bastante gracias a la disponibilidad de estr¾genos de mejor calidad y la posibilidad de utilizar distintas vĒas de administraci¾n.
En las mujeres, la pubertad se puede inducir por distintos mķtodos: estr¾genos conjugados de origen equino, etinilestradiol, 17 beta-estradiol y progestßgenos, entre otros; todos los mķtodos son vßlidos seg·n las condiciones de la paciente y siempre que se disponga de un tiempo de espera para cada una de las etapas, para remedar la pubertad normal. En la actualidad, el etinilestradiol es la forma menos onerosa de inducir la pubertad, de modo que es el mķtodo de elecci¾n en los hospitales del servicio p·blico; tambiķn hay otras formas farmacķuticas, entre ellas cremas, geles y parches, que podrĒan lograr el efecto buscado. Bßsicamente, hay que tener en cuenta que la inducci¾n se debe hacer imitando la pubertad normal fisiol¾gica.
En los ni±os, en el Hospital Roberto del RĒo no hay experiencia en sustituci¾n, sino con testosterona por vĒa parenteral. La masculinizaci¾n con algunos preparados de testosterona administrados por vĒa oral es menos eficiente en cuanto a lograr una buena cantidad de vello masculino, buena masa muscular, etc., de modo que es mßs c¾modo y menos costoso administrar testosterona por vĒa parenteral, como enantato.
┌ltimamente se estß utilizando inhibidores de la aromatasa como tratamiento de la pubertad retrasada. En un estudio doble ciego, controlado con placebo, aleatorio, que incluy¾ a 23 varones con pubertad retrasada constitucional cuya edad media era 15 a±os, sin evidencias de estir¾n puberal a esa fecha, se dividi¾ la muestra en tres grupos:
al primer grupo se le administr¾ enantato de testosterona mßs letrozole; al segundo grupo se le administr¾ enantato de testosterona y placebo y el tercer grupo fue el grupo control, es decir, no recibi¾ tratamiento. En los grupos 2 y 3, que no recibieron inhibidor de la aromatasa, el 17 beta estradiol aument¾, mientras que en el grupo 1 se mantuvo, pero seis meses despuķs de la suspensi¾n del tratamiento, los niveles de estradiol se igualaron en los tres grupos. La testosterona subi¾ en los tres grupos, ya que eran varones en etapa de crecimiento, pero en el grupo 1 el aumento fue mucho mßs exagerado a los cinco meses de tratamiento; al suspender el tratamiento, los niveles de testosterona tambiķn se igualaron en los tres grupos. A los cinco meses de tratamiento, en el grupo 1 aumentaron la LH y la FSH y aument¾ la respuesta de LH a GnRH nativo, mientras que la respuesta de FSH a GnRH no vari¾. Los autores concluyeron que estos resultados demuestran que la inhibici¾n de los estr¾genos end¾genos produce aumento de LH y FSH, a pesar de las altas concentraciones de andr¾genos.
En el estudio aludido tambiķn se midi¾ la inhibina B y se comprob¾ que hubo aumento en el grupo 1. Se midi¾ ademßs el eje somatotr¾fico, hormonas como la IGF-1 y la proteĒna transportadora IGFBF-3, las cuales aumentaron en el grupo 2, que s¾lo estaba recibiendo testosterona y se mantuvieron los niveles constantes en el grupo 1. Al suspender el tratamiento, en el grupo 1 se observ¾ aumento de la velocidad de crecimiento, a los 18 meses de seguimiento; el grupo 2, que s¾lo recibi¾ testosterona, creci¾ solamente durante los cinco primeros meses de tratamiento. Ademßs, se comprob¾ que el letrozole gener¾ un retraso en la maduraci¾n ¾sea; por lo tanto, no hubo aumento de la talla en los pacientes que no fueron tratados con letrozole. Lo anterior sugiere que los andr¾genos administrados en forma aislada no incrementan la talla adulta y confirma lo que se se±al¾ sobre el enantato de testosterona en el crecimiento constitucional. Los resultados anteriores se correlacionaron con una mayor talla estimada para la adultez. De igual forma, se observ¾ aumento de la talla estimada en todos los pacientes del grupo 1, en rango de 2,5 cms a 8,8 cms, excepto en un paciente. En cambio, en los pacientes de los grupos 2 y 3, que no recibieron el inhibidor de la aromatasa, no se modific¾ la talla estimada a los 18 meses de tratamiento (5).
Lo anterior hace suponer que la inhibici¾n de los estr¾genos, en los adolescentes en crecimiento, genera un aumento de la talla final; que no todos los pacientes con pubertad retrasada explotan su probable potencial genķtico de crecimiento; y que ķse es el motivo por el cual la talla final de estos ni±os es algunos centĒmetros menor.
Referencias
- BPract and Res Clin End Met 2002; 16 (1):123-138.
- BPract and Res Clin End Met 2002; 16(1):73-90.
- Horm Res 2003; 60 (suppl 3):35-48.
- Clinical Endocrinology (2003) 58, 267-272.
- Wickman, Lancet 2001; 357:1743- 48.

Esta
obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.









 Estudios originales
Estudios originales