Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
La bacteria Helicobacter pylori llegó al continente americano hace 12.000 ańos (1). A Sudamérica llegó hace aproximadamente 5.400-4.600 ańos A.C. según seńala una investigación de Pelayo Correa, patólogo colombiano, quien encontró Helicobacter en las deposiciones que se encuentran al lado de momias pertenecientes a la cultura Chinchorro y corresponden al norte de Arica en el Océano Pacifico. Es probable, que la bacteria por tanto nos haya acompańado desde esa época hasta ahora. Barry Marshall en el ańo 2005 ganó el premio Nobel con su colega Robin Warren por sus estudios sobre el Helicobacter pylori.
El elemento central en los pacientes infectados con H. pylori es la gastritis superficial crónica. Esta es una infección que por lo general se adquiere en la infancia, antes de los 18 ańos.
Cuando se empezó a tratar de entender cómo se ha convivido tanto tiempo con la bacteria, fue evidente que las primeras miradas intentaron entender lo que era esta gastritis, pues el estómago, a diferencia del colon o del intestino delgado, normalmente no tiene infiltración mononuclear, mientras que en presencia de la bacteria siempre tiene infiltración mononuclear. Se dice que “si hay Helicobacter, hay gastritis; si hay gastritis hay Helicobacter”.
Esto permitió plantear un modelo que intenta explicar el primer componente del sistema inmune que es la inmunidad innata; es decir, inespecífico, no adaptativo, que nos ha acompańado desde siempre. Como teóricamente la bacteria no invade, ésta busca domicilio en la luz del estómago y debe interactuar de alguna manera con el epitelio. Esta es la primera etapa en esa inmunidad innata. Los linfocitos mononucleares están localizados por debajo de la mucosa y la bacteria está en el lumen, y entre ambos se encuentra el epitelio que es donde se produce la primera interacción entre bacteria y huésped.
La bacteria no es capaz de sobrevivir en el ambiente luminal rodeada de ácido clorhídrico, lo rehúye y le es más cómodo un pH neutro. Para alcanzar el pH neutro cercano al epitelio, se debe generar una gradiente desde el lumen al epitelio, y esta gradiente se crea en presencia de ureasa y una capa de gel mucoso. Esas bacterias adyacentes al epitelio son las que están vivas y se están reproduciendo y probablemente son el 2% de la población total; en la medida en que se van muriendo se van liberando al lumen y las bacterias que están en el lumen ya no son viables y adquieren otras características morfologicas.
La bacteria sobrevive gracias a la ureasa, una enzima citosólica que se alimenta de la urea del medio liberada desde el epitelio por medio de los canales de urea. La urea sufre hidrolización lo que permite la sobrevida de la bacteria (2).
Sin embargo, todas las bacterias producen ureasa, por lo tanto esto no explica porqué algunas personas presentan úlceras o cáncer y otras no. Si produce ureasa y la bacteria sobrevive, el segundo aspecto necesario es que la bacteria se pueda adherir al epitelio a traves de adhesinas (3). Existen muchas adhesinas, la más conocida es la Bab-A, que permite que la bacteria se adhiera al epitelio y las proteínas flagelares, responsables de la motilidad bacteriana, ayudan a que la contracción del estómago no las elimine. El problema es que todas las bacterias tienen adhesinas y flagelos, por lo tanto todos los Helicobacter teóricamente se podrían adherir a la mucosa, lo que tampoco explica porqué algunas personas desarrollan úlceras o cáncer.
Se conoce que la bacteria sobrevive, se adhiere y secreta sustancias como proteínas que atraen otras células, dentro de ellas la proteína activadoras de neutrófilos (NAP) (4). Esta proteína que interactúa con un receptor de membrana, activa una serie de seńaléticas, activa cascadas, abre canales de calcio, induce cambios conformacionales y esto produce reclutamiento de neutrófilos y fenómenos inflamatorios. Pero nuevamente, todas las bacterias tienen este tipo de proteína, por lo tanto tampoco explica lo que se ha comentado. Si bien explica la razón por la que la bacteria sobrevive, se mantiene y vive con los seres humanos, no explica necesariamente los pronósticos adversos.
Posteriormente se comenzaron a estudiar otro tipo de factores de virulencia o proteínas, entre las cuales las citotoxinas. A principios de los 90, al preparar un cultivo de bacterias con células in vitro, se podía evidenciar que las células se vacuolizaban; no se encontró la explicación hasta que Tim Cover en Nashville en 1996 pudo darse cuenta que se trataba de una citotoxina que tenía una estructura hexamérica (5). Estudios de ese mismo grupo y de grupos italianos encontraron el gen y se dieron cuenta que tenían alelos, y que de acuerdo a la recombinación de los alelos para la síntesis de estas proteínas era posible obtener distintos pronósticos clínicos.
Así se describió que habrían bacterias que producirían úlceras, s1/m1-m2 y bacterias que no, como la s2/m1-m2. En adición a esto, se dieron cuenta de que la citotoxina no solamente producía vacuolización, sino también producía apoptosis e interactuando con proteínas mitocondriales era capaz de activar factores proinflamatorios. También tenía una acción sobre células T-específicas, lo que sugería que esta citotoxina era capaz de llegar hasta la lámina propia.
A esta citotoxina se le denominó VacA y a los pocos ańos se encontró otra proteína que era altamente antigénica llamada CagA. Esta proteína está sintetizada en un gran pedazo de ADN de la bacteria que se llama el islote de patogenicidad. El islote de patogenicidad es un segmento de aproximadamente 30 genes, cada gen genera una proteína y cada proteína se ensambla en una especie de jeringa. Es un sistema llamado sistema secretor tipo IV (SS-IV), el más complejo es una gran estructura y permite inyectar elementos bacterianos a las células de los humanos.
La proteína CagA forma parte del islote, se secreta y entra a la célula, produce complejos y múltiples caminos; los más importantes son a nivel nuclear, provocando cadenas inflamatorias, activación de sistemas proinflamatorios y también una serie de elementos oncogénicos que ha suscitado gran interés de los investigadores dedicados a estudiar el cáncer gástrico (6).
En un principio se creía que si un paciente tenía bacterias VacA positivas o CagA positivas, serían los pacientes más graves y lo contrario con los pacientes que carecían de estos factores. Así fue durante un par de ańos en especial en el hemisferio norte hasta que comenzaron a aparecer los trabajos asiáticos que mostraban que no existía ninguna correlación. En Chile también se estudió y se encontró que prácticamente entre el 90 y el 100% de los pacientes eran CagA positivos (7). Usando otras técnicas, el grupo de Concepción de Apolinaria García, demostró también 84% de cepas CagA.
Para efectos prácticos la gran mayoría de las cepas presentes en Chile son CagA positivos, por lo que no se explica en absoluto esta diferencia entre úlcera, no úlcera, cáncer, no cáncer. Lo mismo se puede decir acerca de los alelos de Vac. Las diferencias no fueron estadísticamente significativas en los estudios nacionales y no se encontró ninguna relación.
Hasta cierto punto la teoría de las bacterias que producen o no cáncer se ha cuestionado y se empezó a estudiar la posibilidad de que el cáncer no estaría asociado a la presencia de la bacteria. La bacteria siempre ha estado con los seres humanos y probablemente ahora existe enfermedad porque el sistema inmune no sabe comportarse con la bacteria. Al encontrarse la bacteria con el sistema inmune por primera vez en la infancia, los macrófagos que forman parte del sistema reconocen antígenos y luego son capaces de reconocer estas proteínas y otras, activándose. Hay evidencia que muestra que estos macrófagos se activan y producen citoquinas proinflamatorias que liberan especies de oxígeno reactivas dańinas que aumentan la producción local de prostaglandinas, la fibrogénesis, el reclutamiento de mayor cantidad de neutrófilos, mayor cantidad de monocitos y, lo más importante, es que estos macrófagos activados o dendríticas activadas atraen a las células T.
Se refiere a la inmunidad adaptativa en que los linfocitos T tienen que reconocer a la bacteria y aceptarla como extrańa o como propia, es decir que tienen la posibilidad de actuar como células efectoras y la destruyen, o actuar como células tolerantes.
La bacteria vive fuera del epitelio, secreta proteínas, las proteínas interactúan con los macrófagos o con las células dendríticas y estas dendríticas expresan un epítope, es decir, algún antígeno de la bacteria. Un linfocito T helper (Th), Th0 que nunca ha reconocido un antígeno previamente, interactúa con las células mencionadas (macrófagos, dendríticas) y tiene dos opciones. En un medio rico en interleuquina 2 y en interferón gamma, se transforma en una Th1 y este Th1 se caracteriza por un cierto perfil de citoquinas que está dado fundamentalmente por interferón gamma.
Esto pareciera ser lo que sucede en adultos, como lo muestran los modelos experimentales de ratones y primates. Sin embargo, la evidencia muestra que los pacientes infectados, sobre todo si tienen úlcera duodenal, presentan niveles significativamente mayores de citoquinas del espectro Th1 (7).
Existe evidencia que sugiere que si en los nińos hay exceso de interleuquina 4, el Th0 se transforma en un Th2 y produce otro tipo de citoquinas. Esto es importante porque esta respuesta, Th1-Th2 pareciera ser mutuamente excluyente; se observa en nińos y es responsable de la producción de los anticuerpos. Los anticuerpos no son capaces de eliminar la bacteria, sólo sirven para establecer el diagnóstico epidemiológico para estudios de prevalencia, como el que se efectuó en Chile en 2003. Los marcadores serológicos ni siquiera sirven para el seguimiento de los pacientes, ya que estos anticuerpos van a persistir por muchos ańos, tanto IgG, como IgA.
En un estudio con un grupo de nińos, adolescentes y adultos, se evaluó la IgG, IgA, IgM y se pudo observar que las sensibilidades y especificidades eran relativamente aceptables para IgG, relativamente buenas para IgA y bastante malas para IgM. La IgM, a diferencia de en otras infecciones, no desempeńa rol alguno (8). La IgG y la IgA sólo sirven para estudios epidemiologicos ya que persisten mucho tiempo.
La respuesta humoral es una respuesta de tipo Th2, pero la respuesta Th2 es la respuesta que entregan las vacunas. Un estudio demostró que los ratones vacunados producían significativamente más interleuquina 4, que es una citoquina TH2, comparado con los controles. Lo mismo sucede con la interleuquina 10 que se produce en mayor cantidad en ratones vacunados. La respuesta Th2 produce anticuerpos que no sirven para erradicar la infección; las vacunas parecieran producir una respuesta Th2.
En un estudio nuestro con nińos, accidentalmente se comenzó a evidenciar interleuquina 10 y los autores observaron que los nińos infectados producían mucho más interleuquina 10, o sea, Th2; esta diferencia fue menor en la adolescencia y desaparecía en los adultos. La interleuquina 12, una citoquina Th1, era menor en los nińos que en los adultos, y la diferencia se iba reduciendo en la adolescencia y desaparecía hacia la adultez.
Teniendo en cuenta esto, los autores pensaron haber encontrado la respuesta: los nińos no presentan úlceras y no manifiestan enfermedad porque tienen una respuesta Th2 y bastaría con que los “adultos se comportaran como nińos” para dar solución al problema. Se pensó que en algún momento debería ocurrir un cambio en comportamiento de la nińez a la adultez. Se diseńo un estudio en pacientes alérgicos que ya tenían una respuesta Th2 y se pudo ver que en los nińos positivos para Helicobacter pylori había una relación inversa, mientras más alergia, menos bacteria o mientras más bacterias, menos alergia. Cuando se analizó el número de test cutáneos positivos, los que tenían presencia de bacterias, tenían significativamente más test cutáneos positivos y, al compararlos con los resultados, los que tenían úlcera péptica tenían menos porcentaje de test cutáneo positivo, sugiriendo que la alergia protegía de la infección, y que si una persona alérgica se infectaba tenían un mejor pronóstico clínico de la infección. Esto era claro en nińos, pero cuando se evaluó en adultos no se observó lo mismo. Es decir, sólo ocurría en los nińos y esto se puede relacionar con la teoría y la hipótesis de la higiene.
La hipótesis de la higiene sostiene que desde el nacimiento todos los seres humanos tienen tendencia a ser Th2, es decir, responder inmunológicamente de esta manera; las células dendríticas reconocen al alérgeno o al antígeno. En la medida en que el mundo se fue higienizando (vacunas, higiene, agua potable, antibióticos y gastroenterólogos de adultos erradicando bacterias), entonces el intestino se quedó con muy poca exposición bacteriana tradicional que producía respuesta clasicamente tipoTh1, por lo tanto este equilibrio Th1-Th2 se perdió, no porque Th2 haya aumentado o porque haya una epidemia de alergia, sino porque disminuyó la balanza de respuesta Th1 (9).
Por otro lado, en África, los nińos con infecciones por parásitos tienen de nacimiento una respuesta Th2, pero esos mismos nińos africanos están con una alta carga bacteriana de enteropatógenos clásicos, por lo tanto también tienen una respuesta Th1. Estos nińos que presentan muchas infecciones, hacen un buen balance entre infección y alergia. En una modelo altamente higienizado el sistema Th1 y Th2 se bloquea, queda descompensado y aparecen los síntomas de la epidemia de alergia.
Luego aparecieron estudios focalizados en las células T regulatorias y se empezó a describir que estas células comandaban la respuesta final. Esto estaba presente en la literatura de modelos de ratón: si a un ratón normal se le extraían todas las células T (las efectoras - Th1 y Th2 -, las citotóxicas y los linfocitos que producen respuesta inmune) y las T regulatorias, y se le transferían a un ratón sin células T, este ratón volvía a la normalidad porque se restauraba su sistema inmune (10). Pero si a este mismo ratón sólo se le transferían las células Th1 y Th2, o sea, células efectoras, en el ratón se producía enfermedad inflamatoria, básicamente colitis ulcerosa, rechazo de tumor o autoinmunidad.
Si al ratón se le transferían algunas células efectoras y muchas T regulatorias, no solamente restauraba normalidad sino que este ratón se volvía tolerante a todo, incluso a injertos de piel de otro animal. En paralelo existía el antecedente de una enfermedad denominada IPEX, caracterizada por una disfunción inmune, poliendocrinopatía y enteropatía autoinmune ligadas al cromosoma X; las madres, eran asintomáticas, pero poseían escasas células T regulatorias. Al estar ligada al cromosoma X, cuando el nińo recibía el cromosoma de su madre, este nińo se comportaba exactamente igual que ese ratón: desarrollaba enfermedad inflamatoria, básicamente colitis ulcerosa, alergia y enfermedades autoinmunes.
El timo en algún momento producía un grupo de células con un marcador nuclear llamado FOXP3, que corresponde a un factor de transcripción “cabeza de tenedor”, (forkhead box P3), estas células con FOXP3 se activaban, y con un marcador de membrana denominado CD25, eran capaces de bloquear las respuestas efectoras; estás corresponden a las células T regulatorias.
Al principio se especuló que estas células T regulatorias venían del timo y esto es lo que se conoció como una célula regulatoria natural, no se nace con ellas, sino que corresponden al 20% de las células circulan en la sangre (de linfocitos). Tienen el marcador FOXP3 y tienen el marcador de membrana. Con el tiempo se aprendió que había también células en la periferia que en algún momento se transformaban y se inventó el nombre TR1, Th3, CD8 regulatorias, etc. Cada una de estas células regulatorias produce un patrón de citoquinas distintas y funcionan por mecanismos distintos. Algunas de estas T regulatorias detienen el ciclo celular de una célula T efectora a través de una dendrítica; otras T regulatorias producen apoptosis, liberando distintas proteínas; otras T regulatorias bloquean el metabolismo del triptófano de las células dendríticas y por tanto nuevamente producen apoptosis o anergia y así mecanismos similares.
Aun cuando se conoce cómo funciona la respuesta inmune ante Helicobacter pylori, no hay una buena comprensión de la razón por la cual el 90% de las personas no se enferman. Por lo tanto, la mayor parte de los humanos que adquieren la infección en la infancia logran controlar la infección y la regulan. Para probar si existe esa regulación se hicieron estudios en ratones y se podían ver ratones normales, con colonización por bacterias, unidades formadoras de colonias, otros ratones sin T regulatorias y existía una clara diferencia en ausencia de T regulatorias, con menos unidades formadoras de colonias. Asimismo, al observar el ADN bacteriano y la histología de estos ratones sacrificados cuatro semanas después de infectarse, los ratones sin T regulatorias tienen gran inflamación porque hay respuesta Th1 que no tiene contrarregulación, por eso se llaman regulatorias; el ratón normal, en cambio, tiene un equilibrio en su gastritis (11).
Esto también se ha visto en múltiples estudios en los que se pueden ver ejemplos clásicos de experimentos en ratones normales y ratones infectados que tienen linfocitos efectores y ratones infectados que no tienen T regulatorias. En ausencia de T regulatorias tienen una gran inflamación lo que vendría a ser más parecido a un modelo de adulto.
Posteriormente surgía la pregunta de si esto existía en adultos. Hay escasa evidencia con respecto a esto. En la Figura 1 se muestran citometrías de flujo, en los cuadros se marcan las células T regulatorias y también se ven células del antro, del duodeno y de la sangre en personas infectadas y no infectadas de adultos. Existe una diferencia que no se ha analizado estadísticamente, siempre a favor de las personas infectadas. Pareciera que los adultos tienen más T regulatorias cuando están infectados que cuando no están infectados. Tienen más T regulatorias en el antro que los no infectados, en el duodeno y no necesariamente en la sangre.
Figura 1: Citometría de flujo en pacientes adultos.
El trabajo más importante es el de John Atherthon de Nottingham (12) que demostró que adultos infectados expresan significativamente mayor marcadores T regulatorios, esto es por RT-PCR, interleuquina 10, FOXP3, TGF-Beta. Si uno compara es importante la diferencia entre el infectado y el no infectado. Más importante todavía, se estudiaron sólo los infectados y separó los que presentaban úlcera de los que no y se halló que los ulcerosos prácticamente no tenían respuesta T regulatoria, es decir, a mayor respuesta T regulatoria mayor protección.
Un trabajo en el que se compararon nińos chilenos con un grupo control se encontró que nińos infectados y adultos infectados tienen el mismo puntaje de colonización o bien intensidad de carga bacteriana; pero los adultos infectados tienen más de dańo histológico. Sin embargo, cuando se revisó la producción de citoquinas en la mucosa gástrica de estos nińos, los infectados tenían mucha mayor cantidad de marcadores T regulatorias, TGF-beta e interleuquina 10 que los adultos infectados. Se analizaron las biopsias por inmunofluorescencia y se realizó recuento de células FOXP3 mostrando que los nińos infectados no sólo producen mayor cantidad de T regulatorias, sino que probablemente está dado porque tienen un mayor recuento de células FOXP3, sugiriendo que la infección pediátrica efectivamente si tiene una respuesta T regulatoria, y lo que se ha visto en ratones, se reproduce en nińos (13).
Otro ejemplo descrito recientemente es la pueril infección por Enterobious vermicularis, prácticamente sin inflamación histológica, pero lleno de células T regulatorias; versus un adulto con colitis ulcerosa, la inflamación es clásica y hay escasa cantidad de células T regulatorias (14). Por lo tanto es muy probable que este modelo descrito para Helicobacter pylori se podrá ver para una serie de otros fenómenos en los próximos ańos.
En el 2009 se encontró otra célula T helper, denominada 17. Estas eran T regulatorias que venían comprometidas, pero que en el camino se encontraron con interleuquina 6 y se produjo un cambio terminando siendo células Th17. En Chile se evaluó si los pacientes producían interleuquina 17. En un estudio que no ha sido publicado a la fecha de este seminario, se mostró que los adultos chilenos infectados expresan cantidades significativamente mayores de interleuquina 17 comparado con adultos no infectados y con nińos.
Probablemente una célula CD4 nativa en algún momento se va diferenciar a Th1, Th2, Th17 o T regulatoria. Th1 y Th17 probablemente sean similares, Th17 probablemente estuvo presente en la evolución del ser humano mucho antes y probablemente esta respuesta ha existido antes incluso que Th1. Th2 pareciera ser una respuesta descontrolada en ausencia de Th1 y T regulatorias pareciera ser el mecanismo regulador de todo el sistema.
Todo sugiere que lo adultos tienen incrementada la respuesta Th1-Th17 (Figura 2), y que los nińos tienen una sobreexpresión de respuesta T regulatoria, que es capaz de bloquear la respuesta Th1 y probablemente la respuesta Th17 (Figura 3).
Figura 2: Respuesta inmune en adultos.
Figura 3: Respuesta inmune en nińos.
En la actualidad se cree que la respuesta inmune específica contra H. pylori está dada por la balanza que se muestra en la Figura 4, en donde hay normalmente predominio de respuesta Th1, pero que un predominio de respuesta T regulatoria antiinflamatoria es capaz de suprimir la respuesta adaptativa y no manifestarse como enfermedad.
Figura 4: Modelo propuesto para la infección por H. pylori y enfermedades asociadas.
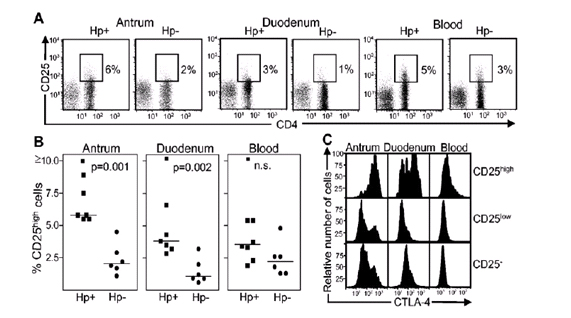 Figura 1: Citometría de flujo en pacientes adultos.
Figura 1: Citometría de flujo en pacientes adultos.
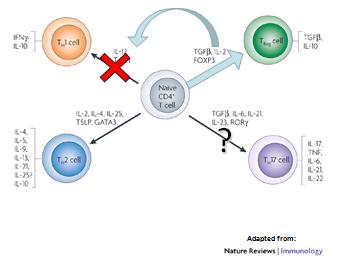 Figura 2: Respuesta inmune en adultos.
Figura 2: Respuesta inmune en adultos.
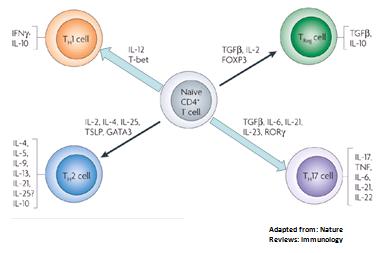 Figura 3: Respuesta inmune en nińos.
Figura 3: Respuesta inmune en nińos.
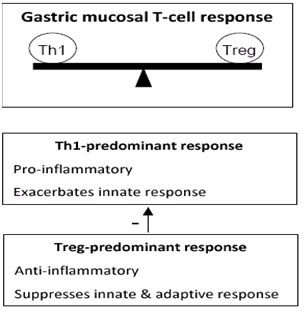 Figura 4: Modelo propuesto para la infección por H. pylori y enfermedades asociadas.
Figura 4: Modelo propuesto para la infección por H. pylori y enfermedades asociadas.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
The bacteria called Helicobacter pylori arrived to the American continent 12,000 years ago (1), reaching South America roughly 5,400-4,600 years AC according to research by Pelayo Correa, a Colombian pathologist who found Helicobacter in stool next to Chinchorro mummies in the North of Arica close to the Pacific Ocean. In 2005, Barry Marshall was awarded the Nobel Prize for his studies on Helicobacter pylori together with Robin Warren.
Autor:
Paul Harris[1]
Citación: Harris P. Immunity and Helicobacter pylori. Medwave 2011;11(03):e4950 doi: 10.5867/medwave.2011.03.4950
Fecha de envío: 3/1/2011
Fecha de aceptación: 14/1/2011
Fecha de publicación: 1/3/2011
Origen: solicitado
Tipo de revisión: sin revisión por pares
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999 May 21;284(5418):1328-33. | CrossRef | PubMed |Mizote T, Yoshiyama H, Nakazawa T. Urease-independent chemotactic responses of Helicobacter pylori to urea, urease inhibitors, and sodium bicarbonate. Infect Immun. 1997 Apr;65(4):1519-21. | PubMed | PMC |Algood HM, Cover TL. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin Microbiol Rev. 2006 Oct;19(4):597-613. | CrossRef | PubMed | PMC |Montecucco C, Rappuoli R. Living dangerously: how Helicobacter pylori survives in the human stomach. Nat Rev Mol Cell Biol. 2001 Jun;2(6):457-66. | CrossRef | PubMed |Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol. 2005 Apr;3(4):320-32. | CrossRef | PubMed |Hatakeyama M. SagA of CagA in Helicobacter pylori pathogenesis. Curr Opin Microbiol. 2008 Feb;11(1):30-7. | CrossRef | PubMed |Serrano C, Diaz MI, Valdivia A, Godoy A, Pena A, Rollan A, et al. Relationship between Helicobacter pylori virulence factors and regulatory cytokines as predictors of clinical outcome. Microbes Infect. 2007 Apr;9(4):428-34. | CrossRef | PubMed |Serrano CA, Gonzaez CG, Rollan AR, Duarte I, Torres J, Pena AJ, et al. Lack of diagnostic utility of specific immunoglobulin M in Helicobacter pylori infection in children. J Pediatr Gastroenterol Nutr. 2008 Nov;47(5):612-7. | CrossRef | PubMed |Yazdanbakhsh M, Kremsner PG, van Ree R. Allergy, parasites, and the hygiene hypothesis. Science. 2002 Apr 19;296(5567):490-4. | CrossRef | PubMed |Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008 May 30;133(5):775-87. | CrossRef | PubMed |Rad R, Brenner L, Bauer S, Schwendy S, Layland L, da Costa CP, et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology. 2006 Aug;131(2):525-37. | CrossRef | PubMed |Robinson K, Kenefeck R, Pidgeon EL, Shakib S, Patel S, Polson RJ, et al. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut. 2008 Oct;57(10):1375-85. | CrossRef | PubMed |






 Estudios originales
Estudios originales