Revista Biomķdica Revisada Por Pares

Este texto completo es una transcripci¾n editada y revisada de una conferencia dictada en el XV Curso de Extensi¾n de PediatrĒa, organizado por el Servicio de PediatrĒa del Hospital ClĒnico San Borja Arriarßn, el Departamento de PediatrĒa Centro de la Facultad de Medicina de la Universidad de Chile, el Servicio de Salud Metropolitano Central y la Direcci¾n de Atenci¾n Primaria. Se realiz¾ entre abril y noviembre de 2006, y sus directores fueron el Dr. Francisco Barrera y la Dra. Cristina Casado.
La epistaxis recurrente es un motivo frecuente de consulta en pediatrĒa. La mayorĒa de los casos son leves y autolimitados, pero hay un peque±o porcentaje de casos graves, que pueden producir anemia secundaria. En la anamnesis del paciente que consulta por epistaxis es muy importante preguntar por los antecedentes personales y familiares de sangrado e investigar en forma dirigida la historia de petequias, p·rpura, equimosis de fßcil aparici¾n, hemorragias quir·rgicas o alveolorragias posteriores a extracciones dentarias, asĒ como antecedentes de sĒndrome de mala absorci¾n y de ingesta de medicamentos antiplaquetarios.
La irrigaci¾n de la nariz proviene de la arteria car¾tida interna y externa, y de sus ramas; por un lado estß la car¾tida interna, con las arterias etmoidales anterior y posterior, que irrigan la parte posterior y superior de la nariz; de la car¾tida externa viene la arteria maxilar interna, cuya rama, la arteria esfenopalatina, irriga la parte posterior e inferior de la nariz, por medio de las arterias nasales. En la porci¾n anterior del tabique nasal (Fig. 1), se originan anastomosis y se forma un verdadero plexo vascular que se conoce como plexo de Kiesselbach: 90% de las epistaxis se originan en esa zona.
Figura 1. Vascularizaci¾n de la nariz.
En cuanto a la etiologĒa de la epistaxis, las causas pueden ser locales o generales (sistķmicas). Entre las causas locales estß la ruptura de vasos de la zona de Little o de Kiesselbach, por sequedad de la mucosa; la desviaci¾n del tabique nasal; traumatismos nasales; la epistaxis digitorum, por grataje de la nariz con los dedos; la presencia de cuerpo extra±o intranasal, que suele ocurrir en los ni±os mßs peque±os; y rinitis virales o bacterianas. Dentro de las causas sistķmicas o generales estßn las discrasias sanguĒneas; la ingesta de Aspirina o antiinflamatorios no esteroidales (AINES); la mielosupresi¾n secundaria a quimioterapia; la hipertensi¾n arterial, mßs bien en personas mayores; y la telangectasia hemorrßgica familiar, que es un cuadro grave. El diagn¾stico de la epistaxis es muy fßcil, porque consiste en una hemorragia externa evidente. La rinoscopĒa anterior, en las epistaxis anteriores, que son mucho mßs frecuentes, es suficiente para hacer el diagn¾stico. Las epistaxis posteriores sangran hacia la rinofaringe, por lo que pueden causar una pseudohematemesis, debido a la sangre deglutida.
El tratamiento local es fundamental, cualquiera sea la causa de la epistaxis. La primera medida es el taponamiento nasal anterior, que se puede hacer con algod¾n a presi¾n, al que se puede agregar un vasoconstrictor, como adrenalina, o un antifibrinolĒtico local, como el ßcido tranexßmico; este tap¾n se debe mantener durante 3 a 7 dĒas, seg·n la cuantĒa de la hemorragia. Si no es suficiente el taponamiento nasal, se puede cauterizar el sitio sangrante con nitrato de plata. En las epistaxis posteriores se hace un taponamiento nasal posterior, que consiste en introducir una sonda Foley por la fosa nasal que sangra hasta llegar a la rinofaringe, donde se infla el bal¾n y se tracciona para que quede enclavado en la coana, lo que determina vasoconstricci¾n de la mucosa. En caso de epistaxis masiva, incluso se puede hacer una ligadura de la arteria maxilar interna y sus ramas.
La epistaxis se debe estudiar cuando es recurrente o masiva, al punto de causar anemia o necesitar transfusi¾n; cuando forma parte de una enfermedad hemorragĒpara general; cuando hay antecedente de trastorno de coagulaci¾n en un miembro de la familia; cuando hay pruebas de coagulaci¾n alteradas en exßmenes preoperatorios; y cuando hay sangrado difuso, sin explicaci¾n, durante una operaci¾n o una extracci¾n dentaria, o despuķs de ella. La etiologĒa de la epistaxis, como todo sangrado mucocutßneo, puede deberse a una trombocitopenia, disfunci¾n plaquetaria, enfermedad de Von Willebrand o telangectasia hemorrßgica hereditaria, que son las causas mßs frecuentes.
La coagulaci¾n se compone de tres partes principales: la hemostasia primaria, la hemostasia secundaria o coagulaci¾n propiamente tal, y el sistema fibrinolitico. La hemostasia primaria (Fig. 2) se pone en marcha cuando se produce una lesi¾n vascular, de modo que se expone el subendotelio; luego, la interacci¾n entre el colßgeno, la fibronectina y el factor de Von Willebrand provoca la adhesi¾n plaquetaria, con lo que la plaqueta cambia de forma: se contrae, liberando serotonina y tromboxano-ADP. La consecuencia de este fen¾meno es la agregaci¾n plaquetaria; posteriormente se forma el trombo plaquetario, que es muy frßgil, y luego viene la segunda etapa, que es la hemostasia secundaria.
Figura 2. Hemostasia primaria.
La hemostasia secundaria tiene una vĒa intrĒnseca y una vĒa extrĒnseca, y ambas tienen una vĒa com·n. En la vĒa intrĒnseca siguen actuando los factores XII, XI y IX, que se activan y luego activan el factor X y lo transforman en X activado. La vĒa extrĒnseca, que puede activarse tambiķn debido a un trauma, tambiķn activa el factor X, por intermedio del factor VII, y ahĒ parte la vĒa com·n. En ķsta, el factor X activa el factor V, el que act·a sobre la protrombina y la transforma en trombina, la que, a su vez, act·a sobre el fibrin¾geno y forma la fibrina. El factor XIII interviene como factor estabilizador de la fibrina y origina una fibrina entrecruzada, mucho mßs firme y eficiente desde el punto de vista hemostßtico.
Figura 3. Hemostasia secundaria.
Por ·ltimo, el sistema fibrinolĒtico permite que se recanalice el vaso sanguĒneo que ha quedado obstruido por el coßgulo. Esta etapa comienza con la activaci¾n del plasmin¾geno plasmßtico, el que asĒ se convierte en plasmina; la acci¾n de esta ·ltima sobre la fibrina causa la aparici¾n de los productos de degradaci¾n de la fibrina (PDF). A su vez, la plasmina queda inactivada por un sistema de antiplasminas compuesto por la alfa 2 antiplasmina y la alfa 2 macroglobulina, lo que permite la recanalizaci¾n del vaso (Fig. 4).
Figura 4. Sistema fibrinolĒtico.
La hemostasia primaria se eval·a mediante el tiempo de sangrĒa, o mķtodo de Ivy, que es una prueba global para medir in vivo la pared vascular; el n·mero de plaquetas, sobre todo cuando es menor de 50.000; la adherencia de las plaquetas al subendotelio; y la agregaci¾n plaquetaria. Consiste en hacer una incisi¾n de 1 cm de largo y 1 mm de profundidad en la cara anterior del antebrazo y medir el tiempo que demora en coagular la sangre. El valor normal es de 3 a 8,5 minutos, pero puede variar de un laboratorio a otro. El tiempo de sangrĒa se altera en presencia de alteraci¾n de la pared vascular; trombocitopenia, en especial menor de 50.000; defecto de adherencia de las plaquetas al subendotelio, como ocurre en la enfermedad de Von Willebrand, uno de los diagn¾sticos diferenciales de la epistaxis; defectos de la secreci¾n plaquetaria; trombocitopatĒas adquiridas por enfermedad hepßtica o renal; fßrmacos, como Aspirina, ticlopidina y clopidogrel.
La funci¾n plaquetaria se estudia mediante agregometrĒa, prueba que permite estudiar la funci¾n plaquetaria in vitro y cuyo resultado se expresa como porcentaje de agregaci¾n. Tambiķn es posible, en algunos laboratorios, estudiar las glicoproteĒnas de membrana de las plaquetas, la cinķtica plaquetaria y la retracci¾n del coßgulo. La hemostasia secundaria, o coagulaci¾n propiamente tal, se puede evaluar mediante la tromboplastina parcial activada (TTPA o tiempo de cefalina), que es el tiempo de coagulaci¾n al a±adir al plasma una sustancia con carga negativa; dicha sustancia es el KaolĒn, que activa la vĒa intrĒnseca. El kaolĒn se altera en caso de dķficit de los factores de la vĒa intrĒnseca VIII, IX, X, XI, XII, precalicreĒna y cinin¾geno de alto peso molecular; y en presencia de heparina y anticoagulante l·pico.
La vĒa extrĒnseca de la coagulaci¾n se eval·a con el tiempo de protrombina de Quick, que es el tiempo de coagulaci¾n al a±adir al plasma tromboplastina (factor tisular, fosfolĒpido, calcio) y que mide la vĒa extrĒnseca, es decir, los factores II, V, VII, X y I. Se alarga en caso de hepatopatĒa y en presencia de anticoagulantes orales; por eso, los tratamientos con anticoagulantes orales se monitorizan con los valores del tiempo de protrombina. El tiempo de trombina es el tiempo de coagulaci¾n obtenido al a±adir al plasma una concentraci¾n baja de trombina; mide la capacidad de polimerizar del fibrin¾geno y es muy sensible a la heparina; se alarga en la hipofibrinogenemia y cuando estßn aumentados los PDF. Tambiķn permite medir los factores, fibrin¾geno, heparina I, II, V, VII y X y los factores VIII, IX, XI, XII.
Otra prueba de coagulaci¾n es la medici¾n del factor Von Willebrand, mediante la actividad del cofactor ristocetina, que se mide con tķcnica de aglutinaci¾n. Para este efecto se utiliza un reactivo que contiene plaquetas humanas estabilizadas, ristocetina y EDTA, y que induce agregaci¾n plaquetaria, mediante uni¾n del factor Von Willebrand a los receptores GPIb ubicados sobre la plaqueta. Otro posible examen es el anßlisis de la composici¾n multimķrica del factor Von Willebrand, que informa sobre la estructura y distribuci¾n de los distintos multĒmeros, pero es una prueba difĒcil, no estß siempre disponible y estß sujeta a diversos factores. Tambiķn se puede hacer detecci¾n de inhibidores.
En la Tabla I se resumen las diferencias entre la epistaxis vascular, que es la causa mßs frecuente en pediatrĒa y que explica 98% a 99% de los casos de epistaxis recurrente, y la enfermedad de Von Willebrand, de baja prevalencia, pero que es el principal diagn¾stico diferencial. La epistaxis vascular es de alta prevalencia en los ni±os, a causa de una fragilidad capilar; da manifestaciones localizadas a nivel nasal; las pruebas de hemostasia son normales; da epistaxis en la edad preescolar y regresa en la pubertad; hay historia familiar de epistaxis recurrente, por lo general en el padre; y el tratamiento clßsico es con vitamina C y citroflavonoides. En cambio, la enfermedad de Von Willebrand, que tambiķn causa epistaxis recurrente, tiene una prevalencia de 0,8 de la poblaci¾n en Chile; su causa es la falta de adherencia de la agregaci¾n plaquetaria y de la coagulaci¾n; da epistaxis recurrentes y hemorragias desproporcionadas durante una cirugĒa o despuķs de ella; es muy frecuente que se manifieste como una hemorragia importante despuķs de una tonsilectomĒa y adenoidectomĒa; el TTPA y el tiempo de sangrĒa estßn prolongados; la epistaxis parte en la edad preescolar y por lo general es permanente, aunque a veces tiende a regresar en la pubertad, porque los estr¾genos aumentan el factor Von Willebrand; y hay historia familiar positiva. El tratamiento dependerß del tipo de enfermedad de Von Willebrand.
Tabla I. Epistaxis vascular y enfermedad de Von Willebrand.
El factor Von Willebrand (FVW) es una plasmoproteĒna completa, grande, compuesta por 2.791 aminoßcidos que forman mon¾meros, dĒmeros y multĒmeros. El gen del FVW se localiza en el cromosoma 12. Este factor tiene dos funciones en la hemostasia: por un lado es mediador de la adherencia plaquetaria al endotelio vascular da±ado, en el cual el FVW se une a los receptores GPIb y GPIIb/GPIIIa de las plaquetas y favorece la formaci¾n del trombo plaquetario, que es la primera fase en la hemostasia primaria; por otro lado, act·a como proteĒna transportadora y estabilizadora del factor VIII, el que act·a en el proceso de la coagulaci¾n y formaci¾n de la fibrina. Gracias al FVW, el factor VIII tiene una sobrevida normal; en ausencia de aquķl, se depura rßpidamente de la sangre. El FVW se sintetiza en las cķlulas endoteliales y los megacariocitos; 85% de lo que se produce se almacena en los grßnulos secretorios de Weibel-Palade, en las cķlulas endoteliales, y 15% se almacena en los grßnulos alfa de las plaquetas.
La enfermedad de Von Willebrand es la mßs frecuente de las enfermedades hemorragĒparas hereditarias; su prevalencia es de 0,8% a 2% de la poblaci¾n; afecta por igual a hombres y mujeres; es de herencia autos¾mica dominante y tiene alto poder de penetraci¾n y gran heterogeneidad dentro de la familia; se caracteriza por la disminuci¾n cuantitativa o cualitativa del FVW plasmßtico y alteraci¾n de la hemostasia primaria y de la coagulaci¾n. En 1926, Eric Von Willebrand describi¾ un raro trastorno hemorragĒparo en una familia que vivĒa en la costa de Finlandia, cuyo cuadro clĒnico constituye la enfermedad que lleva su nombre y que se caracteriza por hemorragia mucocutßnea (80%) y con la epistaxis a repetici¾n como uno de los signos mßs frecuentes; puede haber equimosis (76%), alveolorragia (50%), gingivorragia (47%), hemorragia intraoperatoria o post cirugĒa (60%), menorragia y metrorragia en la mujer.
Este cuadro clĒnico presenta gran variabilidad clĒnica dentro de la misma familia; los sĒntomas no son constantes y hay perĒodos asintomßticos. La mayorĒa de los portadores son sintomßticos leves y no requieren tratamiento especial; y s¾lo 65% de los heterocigotos tienen sĒntomas. El diagn¾stico de la enfermedad de Von Willebrand es complejo y difĒcil; se requiere una buena historia clĒnica, la que no se puede reemplazar con los estudios de laboratorio, si bien ellos sirven como complemento. El tiempo de sangrĒa estß alargado; el estudio de la molķcula del factor VIII demuestra que estß disminuida en la misma proporci¾n que el FVW y puede ser una macromolķcula. En la determinaci¾n antigķnica cuantitativa del FVW se encuentran disminuidas las formas 1 y 3 de este factor. La medici¾n de la actividad del FVW mediante CoF Ristocetina determina la velocidad con que el FVW plasmßtico aglutina las plaquetas normales; su valor se encuentra disminuido en todas las formas de la enfermedad. Otra prueba es la aglutinaci¾n de plaquetas inducidas con ristocetina (APIR), la que se encuentra disminuida en esta enfermedad, pero es poco sensible en las formas leves; sirve en el tipo 2-B plaquetario.
El anßlisis multimķrico del FVW antigķnico no estß disponible en todos los laboratorios, porque exige mucha experiencia, ya que se hace por electroforesis. Ademßs, los valores se pueden alterar debido a una serie de factores, como la edad, el sexo, los ciclos hormonales, el hipo o hipertiroidismo y el embarazo. Tambiķn estß el problema del control genķtico del grupo sanguĒneo: se sabe que el grupo ABO tiene una influencia importante en los niveles de antĒgeno FVW; el grupo O tiene los niveles mßs bajos, con 75 U/dL, seguido por el grupo A, con 106 U/dL; el B, con 117 U/dL; y, finalmente, el grupo AB, con 123 U/dL, lo que dificulta la determinaci¾n de un punto de corte exacto.
La clasificaci¾n de la enfermedad de Von Willebrand se basa en el fenotipo de la enfermedad en plasma y plaquetas. El tipo 1, que es la forma mßs frecuente, es una deficiencia cuantitativa parcial del FVW (80%); el FVW funcional corresponde a 20% a 50% de lo normal. El tipo 2 es una deficiencia cualitativa de la enfermedad de Von Willebrand y tiene 4 subtipos: A, B, M y N. El tipo 3 es la deficiencia completa de factor Von Willebrand; es la forma menos frecuente, pero la mßs grave. De los tipos 2, el 2A es una variante con disminuci¾n de la funci¾n del FVW asociada con ausencia de multĒmeros de alto peso molecular, lo que se asocia con sensibilidad aumentada a la proteolisis. La variante 2B es una variante cualitativa y se caracteriza por aumento de la afinidad del FVW por la GPIb plaquetaria; tambiķn es un Von Willebrand plaquetario. El 2M es una variante con disminuci¾n de la funci¾n del FVW, cuya estructura multimķrica es normal, pero que cualitativamente es anormal. El cuarto subtipo, 2N, es una variante cualitativa con marcada disminuci¾n de la afinidad por el factor VIII C. Es similar a la hemofilia A, por lo que puede inducir a un diagn¾stico err¾neo. En este caso puede dar epistaxis, porque el factor VIII no se puede unir con el punto Von Willebrand y se depura rßpidamente de la circulaci¾n hasta llegar a niveles de 3% a 5%.
El tratamiento de la enfermedad de Von Willebrand exige diagn¾stico de certeza y clasificaci¾n del tipo de enfermedad, ya que a cada uno de los tipos corresponde un tratamiento de elecci¾n y uno de alternativa. El tipo 1, que es el mßs frecuente, se trata con DDAVP (desmopresina), que tendrĒa utilidad; como alternativa se utiliza crioprecipitado o liofilizado. El tipo 2A se trata con crioprecipitado o liofilizado; el tipo 2B, tambiķn con crioprecipitado o liofilizado, pero a veces hay que transfundir plaquetas. El DDAVP puede causar trombopenia. El tipo 3 tambiķn necesita crioprecipitado o liofilizado. En la Tabla II se resume lo expuesto.
Tabla II: Tratamiento de la enfermedad de Von Willebrand, seg·n tipo.
La DDAVP (1-deamino-8-D-arginina vasopresina) es un anßlogo sintķtico de la vasopresina, que aumenta el factor VIII y el FVW; hay un preparado parenteral endovenoso y un spray nasal. La dosis es de 0,3 a 0,4 ug/kg endovenoso en el Von Willebrand tipo 1; en la preparaci¾n nasal se administran 150 ug, lo que equivale a un puff nasal para pacientes de menos de 50 kg. El efecto mßximo se obtiene en 30 a 60 minutos: acorta el tiempo de sangrĒa y disminuye la hemorragia; puede tener efectos colaterales, por ejemplo cefaleas, taquicardia, aumento de la presi¾n arterial, convulsiones, aunque son raras, y rubicundez facial.
El crioprecipitado contiene 100 U de factor VIII; 100 U de FVW; 250 mg de fibrin¾geno; factor XIII y fibronectina. El FVW que contiene el crioprecipitado es de composici¾n multimķrica adecuada, por eso es de primera elecci¾n, en relaci¾n con el liofilizado. La dosis generalmente es una bolsa por cada 10 kg de peso o 25 U de factor VIII por kg de peso, pero depende de la hemostasia que se necesite: para una cirugĒa peque±a esto es suficiente, aunque se puede repetir cada 6 u 8 horas; pero si es una gran cirugĒa la dosis deberß ser mayor. Para calcularla, se multiplica el porcentaje de FVW que se desea obtener por el peso de paciente dividido por 2. Por ejemplo: si se desea obtener 40% del factor, se multiplica 40 por el peso y se divide por 2 y el resultado es la cantidad de FVW que se requiere para conseguir la hemostasia. El liofilizado contiene principalmente factor VIII, con poco FVW. Existe un preparado, que todavĒa no habrĒa llegado a Chile, de nombre comercial Humate-P, que tiene FVW adecuado, con los multĒmeros conservados y se utiliza en dosis de 10 U/kg de peso endovenoso, seg·n la hemostasia que se requiera.
El tratamiento de la enfermedad de Von Willebrand sigue siendo empĒrico y controvertido, ya que no hay correlaci¾n entre la clĒnica y el laboratorio que permita monitorizar la terapia de sustituci¾n, especialmente en el Von Willebrand leve y moderado. En el Von Willebrand grave es mßs fßcil hacer el diagn¾stico, pero se necesita una buena historia, la que no se puede reemplazar con exßmenes del laboratorio, porque, por un lado, se necesita un laboratorio muy experimentado que haga todas las pruebas y, por otro lado, hay que saber interpretarlas de acuerdo con una serie de factores que pueden modificarlas. Incluso una venopunci¾n muy difĒcil puede desencadenar la cascada de la coagulaci¾n, de modo que es difĒcil saber interpretar estos valores. Por eso es que la historia tiene mucha importancia. La terapia antifibrinolĒtica es ·til como coadyuvante, ya que previene la lisis del coßgulo despuķs de que se alcanz¾ hemostasia con DDAVP o crioprecipitado. Los preparados que se utilizan son el ßcido aminocaproico (EACA o Amicar), en dosis inicial de 100 mg/kg, seguida de 50 mg/kg cada 6 horas por 4 a 5 dĒas, hasta detener la hemorragia; y el ßcido tranexßmico (Espercil), en dosis de 50 mg/kg/dĒa endovenoso, fraccionado cada 6 a 8 horas.
El SĒndrome de Bernard-Soulier tambiķn puede dar epistaxis recurrente. Es una enfermedad hemorragĒpara congķnita poco frecuente, de herencia autos¾mica recesiva, causada por la ausencia de GP1b en la membrana plaquetaria. Se manifiesta por trombocitopenia leve y plaquetas gigantes al frotis, con sobrevida disminuida, tiempo de sangrĒa prolongado y aglutinaci¾n plaquetaria con ristocetina, defectuosa. El tratamiento consiste en transfusi¾n de concentrado de plaquetas en casos especiales, pero con cuidado, porque, como estas plaquetas tienen GP1b, pueden desencadenar la producci¾n de isoanticuerpos, lo que a la larga determina que el paciente se vuelva refractario a la transfusi¾n plaquetaria.
La Tromboastenia de Glanzman es una enfermedad hemorragĒpara congķnita, tambiķn rara, en la que hay disfunci¾n plaquetaria, la que consiste en una falla de la agregaci¾n de las plaquetas con los agonistas normales: ADP, colßgeno, ristocetina, etc. Ademßs, hay ausencia de GPIIb/IIIa, que son receptores de fibrin¾geno. El recuento de plaquetas y la morfologĒa son normales y el diagn¾stico se hace por ausencia de agregaci¾n plaquetaria, con todos los agonistas, y ausencia de retracci¾n del coßgulo. El tratamiento tambiķn consiste en transfusi¾n de plaquetas en casos graves y tambiķn existe el riesgo de que se generen isoanticuerpos y refractariedad a la transfusi¾n plaquetaria.

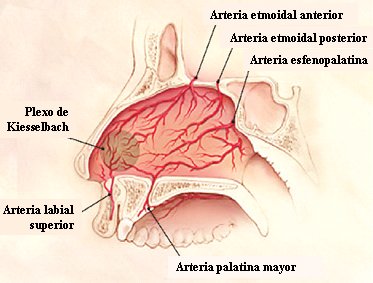 Figura 1. Vascularizaci¾n de la nariz.
Figura 1. Vascularizaci¾n de la nariz.
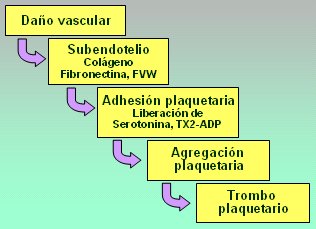 Figura 2. Hemostasia primaria.
Figura 2. Hemostasia primaria.
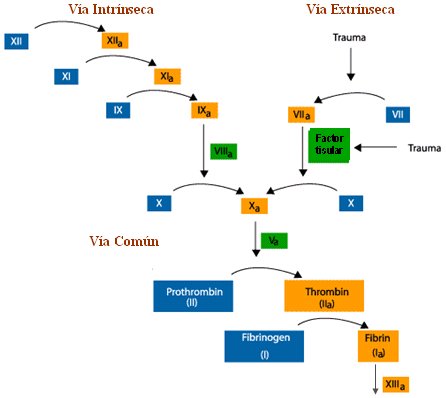 Figura 3. Hemostasia secundaria.
Figura 3. Hemostasia secundaria.
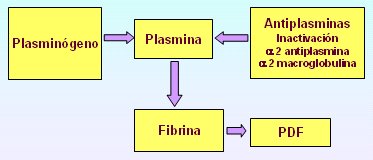 Figura 4. Sistema fibrinolĒtico.
Figura 4. Sistema fibrinolĒtico.
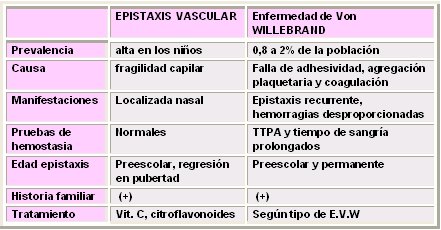 Tabla I. Epistaxis vascular y enfermedad de Von Willebrand.
Tabla I. Epistaxis vascular y enfermedad de Von Willebrand.
 Tabla II: Tratamiento de la enfermedad de Von Willebrand, seg·n tipo.
Tabla II: Tratamiento de la enfermedad de Von Willebrand, seg·n tipo.
 Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Este texto completo es una transcripci¾n editada y revisada de una conferencia dictada en el XV Curso de Extensi¾n de PediatrĒa, organizado por el Servicio de PediatrĒa del Hospital ClĒnico San Borja Arriarßn, el Departamento de PediatrĒa Centro de la Facultad de Medicina de la Universidad de Chile, el Servicio de Salud Metropolitano Central y la Direcci¾n de Atenci¾n Primaria. Se realiz¾ entre abril y noviembre de 2006, y sus directores fueron el Dr. Francisco Barrera y la Dra. Cristina Casado.
Expositora:
Irina Ocheretin P.[1]
Citaci¾n: Ocheretin I. Epistaxis: when to consider further work-up?. Medwave 2007 Ene;7(1):e1981 doi: 10.5867/medwave.2007.01.1981
Fecha de publicaci¾n: 1/1/2007
Nos complace que usted tenga interķs en comentar uno de nuestros artĒculos. Su comentario serß publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la direcci¾n editorial considera que su comentario es: ofensivo en alg·n sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas polĒticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisi¾n por pares.
A·n no hay comentarios en este artĒculo.
Para comentar debe iniciar sesi¾n







 Estudios originales
Estudios originales