Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Palabras clave: linear IgA bullous dermatosis, bullous disease
En este texto se presenta el caso de una paciente de sesenta y cinco ańos con sintomatología de dos meses de evolución consistente en prurito y lesiones hiperpigmentadas anulares en tronco, glúteos y extremidades superiores. En el área flexora de las muńecas presenta vesículas sobre base eritematosa y piel sana, sin evidencia de compromiso mucoso. Al estudio histológico se constata dermatitis vesicular subepidérmica con infiltrado inflamatorio de neutrófilos y eosinófilos. La inmunofluorescencia directa muestra depósito lineal y continuo de inmunoglobulina A en zona de membrana basal, compatible con dermatosis por inmunoglobulina A lineal.
La dermatosis por inmunoglobulina A lineal es un trastorno autoinmunitario poco frecuente, que se caracteriza por la presencia de lesiones vesiculosas y ampollares. Su diagnóstico se confirma con la realización de inmunofluorescencia directa, que muestra la presencia de depósitos de inmunoglobulina A en la unión dermoepidérmica [1]. Su incidencia en Sudamérica aún se desconoce.
A continuación exponemos el presente caso clínico, debido a la baja incidencia de esta patología en nuestra población y a su amplio diagnóstico diferencial, tanto en población pediátrica como adulta. Además, presenta una conducta terapéutica específica en comparación a otras dermatosis ampollares autoinmunes.
Presentación de caso clínico
Paciente femenino de 65 años de edad, con antecedentes de adenoma suprarrenal, hipertensión arterial y enfermedad pulmonar obstructiva crónica secundaria a tabaquismo en tratamiento con atenolol. Refiere cuadro de dos meses de evolución, caracterizado inicialmente por prurito intenso y posterior aparición de lesiones eritematosas asociadas a vesículas, en tronco y extremidades superiores. Éstas aumentaron de tamaño en forma progresiva, adquiriendo algunas una distribución anular y que posteriormente evolucionaron a costras periféricas.
Al examen físico destacó extensa placa hiperpigmentada anular en tronco (Figura 1), de forma irregular, con borde eritematoso y erosiones costrosas de predominio periférico. Presentó lesiones de características similares de menor tamaño en dorso, glúteos (Figura 2) y extremidades superiores. En superficies flexoras de ambas muñecas se asoció a ampollas y vesículas tensas, de contenido seroso, sobre base eritematosa y piel sana (Figura 3). No se observó compromiso de mucosas.
Debido a estos antecedentes clínicos, se planteó como hipótesis diagnóstica dermatosis inmunoglobulina A lineal, por lo que se planificó biopsia incisional para su confirmación. Se comenzó a suministrar 30 milígramos de prednisona por vía oral, después de la obtención de la muestra.
El estudio histológico con tinción de hematoxilina-eosina, evidenció dermatitis vesicular subepidérmica con infiltrado inflamatorio de neutrófilos y eosinófilos (Figura 4), concordante con enfermedad ampollar autoinmune subepidérmica. La inmunofluorescencia directa mostró depósito lineal y continuo de inmunoglobulina A en zona de membrana basal (Figura 5), compatible con dermatosis por inmunoglobulina A lineal.
Examenes de control en rangos de normalidad y actividad de glucosa 6 fosfato deshidrogenasa normal. Se inició tratamiento con 50 miligramos y 100 miligramos de dapsona día por medio, evolucionando con hiperpigmentación post inflamatoria asimétrica en dorso, flanco y axila derecha, sin nuevas lesiones ampollares. Por este motivo, se redujo progresivamente la dosis de prednisona oral hasta la suspensión y se disminuyó dosis de dapsona a 50mg al día, con respuesta clínica favorable.
Figura 1. Gran placa hiperpigmentada anular con costras y eritema periféricos en tronco.
Figura 2. Placa hiperpigmentada anular con costra y eritema periféricos en glúteos.
Figura 3. Vesículas, ampollas y costras en cara anterior distal de antebrazo izquierdo.
La dermatosis por inmunoglobulina A lineal es una patología ampollar crónica, adquirida y de etiología autoinmune [1]. Se caracteriza por la presencia de ampollas subepidérmicas y depósito lineal de autoanticuerpos de inmunoglobulina A, contra múltiples antígenos localizados en la membrana basal. Éstos se visualizan mediante inmunofluorescencia directa o inmunofluorescencia indirecta [2]. Se presenta tanto en población pediátrica como adulta, con algunas diferencias semiológicas [3].
De acuerdo al sitio de división, se describen dos formas de dermatosis por inmunoglobulina A lineal:
La dermatosis por inmunoglobulina A lineal fue reconocida por primera vez como entidad clínica en 1979 por Chorzelski y Jablonska [6]. Presenta una incidencia variable según ubicación geográfica, siendo mayor en China, sureste de Asia y África [1]. En Europa se estima en 0,5 casos nuevos por millón de habitantes al año [7]. La incidencia en América del Sur aún es desconocida.
Puede presentarse a cualquier edad, pero clásicamente muestra dos puntos máximos. El primero en niños entre cuatro y cinco años; y el segundo, como ocurre en el caso presentado, en adultos mayores de 60 años, con leve predominio por el sexo femenino [8].
La mayoría de las investigaciones sobre la enfermedad han mostrado que existe una fuerte asociación entre la dermatosis por inmunoglobulina A lineal y los antígenos leucocitarios humanos B8, CW7 y DR3 [9]. Ello se relacionaría con un inicio precoz de la enfermedad. Por otro lado, se ha demostrado una asociación con el factor de necrosis tumoral-2, cuya presencia se relaciona con un peor pronóstico [10].
El mecanismo fisiopatológico que desencadena la respuesta autoinmune en la dermatosis por inmunoglobulina A lineal, aún es desconocido. Las investigaciones durante los últimos 30 años han identificado diferentes antígenos relacionados, los que se encuentran localizados en la zona de la membrana basal epitelial [11]. En la patogenia de la enfermedad parecen estar involucradas tanto la respuesta humoral como celular [2].
La respuesta humoral está determinada por la producción patológica de inmunoglobulina A contra antígenos de la zona de membrana basal. La mayoría desarrolla anticuerpos contra más de un antígeno, siendo los más comunes el LAD-1 y LABD97, derivados del BPAg2 o colágeno XVII [12],[13]. Una parte del segmento extracelular del BPAg2 se desprende fisiológicamente y es degradado por acción proteolítica para producir fragmentos y nuevos epítopes, proceso conocido como “difusión de epítope” [14]. En modelos in vitro, la unión de anticuerpos inmunoglobulina A causó daño tisular de piel en cultivo y unión de neutrófilos a la membrana basal [15]. La transferencia pasiva de suero en ratones con dermatosis por inmunoglobulina A lineal a un ratón con inmunodeficiencia severa combinada, es capaz de promover infiltración neutrofílica y vesiculación de la membrana basal [16].
En cuanto a la respuesta inmune celular, los análisis inmunohistoquímicos han demostrado la presencia de neutrófilos y eosinófilos [6] cuya activación provocaría el reclutamiento y activación de un mayor número de células polimorfonucleares. Estas serían responsables de liberar citoquinas relacionadas con la disrupción de la membrana basal. El tipo de citoquinas es variado, pero se ha identificado un aumento en la producción de interleuquina 2 mediado por linfocitos T helper 1 y de interleuquina 5 mediado por linfocitos T helper 2 [17].
En relación a la clínica, se presentan diferencias según la edad del paciente:
Dermatosis por inmunoglobulina A lineal en población pediátrica o enfermedad bulosa crónica de la infancia: es la enfermedad ampollar autoinmune adquirida más común de la niñez. Clásicamente se presenta como un episodio agudo de erupción vesicular o ampollar, con compromiso de cara (generalmente peribucal y periocular) y la zona anogenital, incluyendo el abdomen bajo, vulva, muslos, zona perineal y glúteos [18]. Puede existir compromiso mucoso con úlceras o erosiones orales, sangrado nasal y conjuntivitis, pero en menor frecuencia que en población adulta [19].
Se pueden presentar sobre piel sana o en placas eritematosas, las que frecuentemente adoptan un patrón anular o policíclico [20], con ampollas alrededor del borde produciendo los signos de la “corona de perlas” en lesiones circulares, y el “collar de perlas” en trayectos más serpiginosos [7]. Se asocia a síntomas variables, desde prurito a ardor severo.
Esta presentación suele ser autolimitada y remitir en meses o años. Sin embargo, en algunos casos puede persistir en la adultez, con las mismas características clínicas que la forma adulta [21].
Dermatosis por inmunoglobulina A lineal en adultos: la presentación clínica clásica en adultos es compatible con la del caso expuesto. La enfermedad suele comenzar de forma abrupta, con sensación urente variable o prurito intenso asociado a signos de grataje. Evoluciona con vesículas o ampollas claras y/o hemorrágicas, que aparecen en piel normal o con base eritematosa o urticarial. Generalmente son tensas, de distinto tamaño y tienden a formar placas anulares o policíclicas debido a la coalescencia de las lesiones [9]. En adultos se comprometen preferentemente superficies extensoras como tronco, glúteos y cara, especialmente la zona perioral, pudiendo o no ser simétrico [1].
En la paciente presentada no se observó compromiso mucoso, pero en la literatura se describe hasta en el 50% de los pacientes [22] con úlceras y erosiones dolorosas en la cavidad oral, siendo más afectados el paladar blando y duro, los arcos palatinos y la mucosa bucal. Menos frecuente es el compromiso de la lengua, encías, vestíbulo oral y labios [19]. El compromiso nasal y ocular puede ser uni o bilateral, con conjuntivitis crónica que puede terminar en fibrosis y formación de simbléfaron, los que sin tratamiento pueden evolucionar a ceguera [23]. Aunque es poco común, el compromiso de mucosa puede ser el único signo clínico o bien preceder a las lesiones cutáneas [20]. Los genitales se afectan con menor frecuencia que en la población pediátrica.
Si bien la mayoría de los casos de dermatosis por inmunoglobulina A lineal son idiopáticos, como se presume en el caso clínico descrito, se han identificado algunos factores precipitantes en la literatura, tales como:
Drogas: la dermatosis por inmunoglobulina A lineal inducida por drogas, normalmente es autolimitada y suele resolverse rápida y espontáneamente luego de la suspensión del medicamento. La relación temporal con la droga y la rara presencia de autoanticuerpos inmunoglobulina A anti membrana basal, suelen ser útiles en el diagnóstico [2]. La vancomicina es el fármaco que más se ha relacionado con la enfermedad, seguido por amiodarona, antiinflamatorios no esteroidales, captopril, ceftriaxona y metronidazol [1]. El mecanismo de la dermatosis por inmunoglobulina A lineal inducida por drogas es desconocido, posiblemente los fármacos podrían presentar reacción cruzada con autoantígenos de la membrana basal o desenmascarar antígenos ocultos previamente.
Enfermedades sistémicas: algunas enfermedades linfoproliferativas se han asociado a la dermatosis por inmunoglobulina A lineal, como el linfoma no Hodgkin y la leucemia crónica linfocítica [24].
Traumatismos cutáneos: lesiones cutáneas como las quemaduras o la exposición a luz ultravioleta han sido asociadas a la aparición de la enfermedad [25].
Enfermedades gastrointestinales: la colitis ulcerosa es la enfermedad no maligna más comúnmente involucrada, seguida por el lupus eritematoso sistémico y la enfermedad de Crohn [26].
Por último, durante el embarazo suele mejorar la condición clínica, probablemente por una mayor glicosilación de inmunoglobulina A debida a las hormonas del embarazo, alterando la capacidad de unirse al antígeno [27].
Dada la dificultad de diferenciar la dermatosis por inmunoglobulina A lineal de otras enfermedades ampollares por la clínica y los hallazgos histopatológicos, el diagnóstico debe ser confirmado por inmunofluorescencia directa [2]. En todos los casos se debe descartar cuadros inducidos por fármacos [28].
La histopatología se caracteriza por la presencia de ampollas subepitelales con infiltrado predominantemente neutrofílico en la dermis superior, formando microabscesos papilares [29]. Además, se observan eosinófilos y mononucleares [30].
En el caso clínico presentado se realiza inmunofluorescencia directa en muestra de piel perilesional, lo que se considera el estándar de oro diagnóstico [1]. Esto incluye la presencia de depósitos de inmunoglobulina A, a lo largo de la zona de membrana basal en un patrón lineal y continuo, que es patognomónico de la enfermedad. En estos casos, es menos común la presencia de inmunoglobulina G, inmunoglobulina M y complemento C3 [2]. La inmunofluorescencia indirecta es más frecuentemente positiva en población pediátrica (75%) que en adultos (30%) [31].
Mediante la técnica salt split skin puede aumentar la sensibilidad de la serología, observándose principalmente depósito a nivel epidérmico (dermatosis por inmunoglobulina A lineal tipo lámina lúcida) o dérmico (dermatosis por inmunoglobulina A lineal tipo sublámina densa). Sin embargo, en algunos casos se presenta una distribución mixta [16],[32]. Curiosamente, en la mayoría de los casos de dermatosis por inmunoglobulina A lineal inducida por fármacos, no son detectados estos autoanticuerpos contra la zona de membrana basal [33].
La microscopia inmunoelectrónica tiene la capacidad de detectar la localización exacta de los depósitos de complejos inmunes, ya sea en las láminas lúcida, densa o en ambas. Este hallazgo inmunopatológico corresponde a una característica particular de los pacientes con dermatosis por inmunoglobulina A lineal, dando la apariencia de imagen en espejo [34]. Si ninguna de las pruebas es concluyente, immunoblot puede constituir una prueba adicional para identificar el antígeno [35].
El diagnóstico diferencial generalmente se da con otras enfermedades ampollares, según la edad de presentación [1] (Tabla 1).
Tabla 1. Enfermedades ampollares según la edad de presentación.
La dermatosis por inmunoglobulina A lineal requiere de un manejo complejo y multidisciplinario. En la literatura la dapsona es considerada el tratamiento de primera línea, mientras que los corticoides se reservan para casos de difícil manejo [36].
El mecanismo de acción por el cual la dapsona inhibe la quimiotaxis de neutrófilos en el sitio de depósito de inmunoglobulina A no se comprende en la actualidad. Se ha demostrado que inhibe la actividad lisosomas de neutrófilos y la yodación mediada por mieloperoxidasa, pero no tendría efecto sobre los depósitos de anticuerpos o complementos [37]. Por otro lado, la dapsona inhibe la adhesión de neutrófilos a la zona de membrana basal epitelial siendo un efecto dosis dependiente [37].
La dosis inicial es de 25 a 50 milígramos diarios en adultos y 0,5 milígramos por kilo al día en niños, aumentando lentamente hasta un máximo recomendado de 2,5 a 3 milígramos por kilo al día [2], con una dosis media de 100 milígramos para controlar la enfermedad. La respuesta se puede presentar desde 24 hasta 48 horas, con resolución de las lesiones dentro de los días posteriores. Una vez alcanzado el efecto, se recomienda el descenso progresivo y escalonado del medicamento (12,5 a 25 milígramos cada una a dos semanas). Ante la aparición de nuevas lesiones pequeñas, se pueden asociar corticoides tópicos de alta potencia. Se deben evaluar los niveles de glucosa 6 fosfato deshidrogenasa, previo al inicio del tratamiento para evitar que el estrés oxidativo de la dapsona no induzca una crisis hemolítica. Una vez iniciado el tratamiento, se debe controlar con hemograma cada dos a cuatro semanas durante los primeros tres meses, para evaluar la presencia de leucopenia o hemólisis.
La segunda línea de tratamiento consiste en sulfonamidas como sulfapiridina (16 a 60 milígramos por kilo al día), sulfasalazina o sulfametoxipiridazina, sola o en asociación con dapsona. La combinación entrega una eficacia acumulada sin mayor toxicidad [38].
En casos de respuesta parcial se puede asociar a corticoides tópicos o sistémicos [39]. Se ha visto que los corticoesteroides tópicos en piel o mucosa, pueden ser muy efectivos como agentes únicos en casos leves o como coadyuvantes a la terapia sistémica en enfermedad severa [2].
En aquellos pacientes en que la enfermedad no se controla, se pueden usar inmunosupresores como micofenolato, colchicina, ciclofosfamida, ciclosporina o tracolimus tópico [40]. Algunos agentes antimicrobianos como la oxacilina, dicloxacilina, eritromicina, flucloxacilina y cotrimoxazol han demostrado ser efectivos en el tratamiento [41]. El uso conjunto de tetraciclina y niacinamida ha mostrado eficacia en el tratamiento de dermatosis por inmunoglobulina A lineal en distintas series [42],[43],[44]. La inmunoglobulina intravenosa se ha utilizado con éxito en pacientes refractarios, portadores de falla renal crónica o con compromiso ocular crónico [45],[46],[47]. Recientemente se reportó el uso de rituximab en un caso refractario, con respuesta clínica favorable [48]. La mayoría de los pacientes responden rápidamente a la terapia inicial, por lo que estas alternativas se reservan en casos refractarios o con contraindicaciones para su uso.
La terapia sistémica se debe continuar hasta el que el paciente entre en remisión clínica completa, con una dosis de mantención según las características individuales de cada paciente.. Si la enfermedad recurre, se debe reiniciar terapia sistémica y mantenerse por semanas o meses luego de controladas las lesiones [2].
En relación al pronóstico, la dermatosis por inmunoglobulina A lineal infantil es autolimitada, con un promedio de duración entre uno y cinco años [18]. La forma adulta es más crónica y refractaria [2]. El compromiso mucoso puede generar importantes consecuencias funcionales como ceguera [49]. En los casos inducidos por drogas, la resolución comienza en pocos días o hasta semanas después de la suspensión del fármaco [28].
La dermatosis por inmunoglobulina A lineal es una enfermedad inmunomediada poco frecuente, caracterizada por la presencia de depósitos de inmunoglobulina A en la zona de la membrana basal, visible con la inmunofluorescencia directa. Clínicamente puede dividirse en la forma adulta o pediátrica. A pesar de que su forma idiopática es la más frecuente, siempre se debe descartar la presencia de factores desencadenantes como fármacos o enfermedades malignas. El tratamiento de elección es la dapsona, existiendo numerosas alternativas entre las que se encuentran las sulfas, los corticoides y los inmunosupresores. El pronóstico suele ser favorable.
Creemos importante presentar este caso clínico debido a la baja frecuencia de esta patología en Chile, su presentación en distintos grupos etarios y tener un enfoque terapéutico específico.
Agradecimientos
Los autores agradecen al Departamento Dermatología, Hospital Clínico Universidad de Chile por la colaboración prestada para la elaboración de este manuscrito.
Aspectos éticos
El consentimiento informado solicitado por Medwave, ha sido firmado por el paciente; una copia de este fue remitido a la dirección editorial de la revista.
Conflictos de intereses
Los autores han completado el formulario de declaración de conflictos de intereses del ICMJE, y declaran no haber recibido financiamiento para la realización del reporte; no tener relaciones financieras con organizaciones que podrían tener intereses en el artículo publicado, en los últimos tres años; y no tener otras relaciones o actividades que podrían influir sobre el artículo publicado. Los formularios pueden ser solicitados contactando al autor responsable o a la dirección editorial de la Revista.
Financiamiento
Los autores declaran que no hubo fuentes de financiación externas.
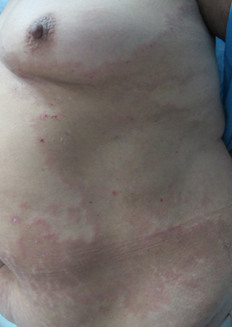 Figura 1. Gran placa hiperpigmentada anular con costras y eritema periféricos en tronco.
Figura 1. Gran placa hiperpigmentada anular con costras y eritema periféricos en tronco.
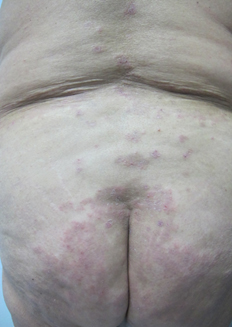 Figura 2. Placa hiperpigmentada anular con costra y eritema periféricos en glúteos.
Figura 2. Placa hiperpigmentada anular con costra y eritema periféricos en glúteos.
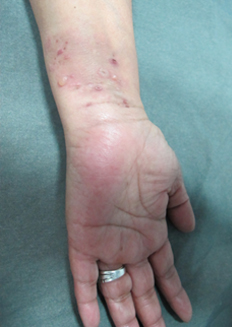 Figura 3. Vesículas, ampollas y costras en cara anterior distal de antebrazo izquierdo.
Figura 3. Vesículas, ampollas y costras en cara anterior distal de antebrazo izquierdo.
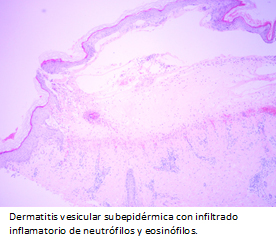 Figura 4. Figura 4. Microscopía óptica. Tinción hematoxilina-eosina 4x: dermatitis vesicular subepidérmica con infiltrado inflamatorio de neutrófilos y eosinófilos.
Figura 4. Figura 4. Microscopía óptica. Tinción hematoxilina-eosina 4x: dermatitis vesicular subepidérmica con infiltrado inflamatorio de neutrófilos y eosinófilos.
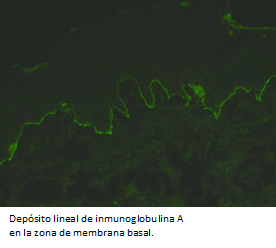 Figura 5. Inmunofluorescencia directa 4x: depósito lineal de Inmunoglobulina A en la zona de membrana basal.
Figura 5. Inmunofluorescencia directa 4x: depósito lineal de Inmunoglobulina A en la zona de membrana basal.
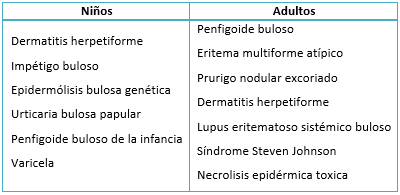 Tabla 1. Enfermedades ampollares según la edad de presentación.
Tabla 1. Enfermedades ampollares según la edad de presentación.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
We present the case of a sixty five year old woman with two months history of pruritus and hyperpigmented annular lesions on the trunk, buttocks and upper extremities. In addition, she presents vesicles with healthy skin on the basis, in the flexor aspect of wrists. No evidence of mucosal involvement. Histological study showed subepidermal vesicular dermatitis with inflammatory infiltrate of neutrophils and eosinophils. Direct immunofluorescence evidenced linear and continuous deposition of immunoglobulin A in basement membrane zone, compatible with linear immunoglobulin A disease.
Autores:
Fernando Valenzuela Ahumada[1], Roberto Bustos Macaya[1], Gabriela Paz Romero Morgado[1], Margarita Sánchez Chacón[1]
Citación: Valenzuela Ahumada F, Bustos Macaya R, Romero Morgado GP, Sánchez Chacón M. Linear immunoglobulin A dermatosis: A case report. Medwave 2017 Abr;17(3):e6901 doi: 10.5867/medwave.2017.03.6901
Fecha de envío: 8/11/2016
Fecha de aceptación: 26/1/2017
Fecha de publicación: 4/4/2017
Origen: no solicitado
Tipo de revisión: con revisión por tres pares revisores externos, a doble ciego
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Venning VA. Linear IgA disease: clinical presentation, diagnosis, and pathogenesis. Immunol Allergy Clin North Am. 2012 May;32(2):245-53, vi. | CrossRef | PubMed |Fortuna G, Marinkovich MP. Linear immunoglobulin A bullous dermatosis. Clin Dermatol. 2012 Jan-Feb;30(1):38-50. | CrossRef | PubMed |Sandoval M, Farias MM, Gonzalez S. Linear IgA bullous dermatosis: report of five cases in Chile. Int J Dermatol. 2012 Nov;51(11):1303-6. | CrossRef | PubMed |Zone JJ, Taylor TB, Kadunce DP, Meyer LJ. Identification of the cutaneous basement membrane zone antigen and isolation of antibody in linear immunoglobulin A bullous dermatosis. J Clin Invest. 1990 Mar;85(3):812-20. | PubMed |Vodegel RM, de Jong MC, Pas HH, Jonkman MF. IgA-mediated epidermolysis bullosa acquisita: two cases and review of the literature. J Am Acad Dermatol. 2002 Dec;47(6):919-25. | PubMed |Conleth A, Zone J. Linear IgA bullous dermatosis. Int J Dermatol. 1999 Nov;38(11):818-27. | PubMed |Fuentelsaz V, Campos M. Dermatosis IgA lineal de la infancia. Rev Pediatr Aten Primaria. 2013;15:141-5. | Link |Chen S, Mattei P, Fischer M, Gay JD, Milner SM, Price LA. Linear IgA bullous dermatosis. Eplasty. 2013 Jul 2;13:ic49. | PubMed |Ingen-Housz-Oro S. [Linear IgA bullous dermatosis: a review]. Ann Dermatol Venereol. 2011 Mar;138(3):214-20. | CrossRef | PubMed |Collier PM, Wojnarowska F, Welsh K, McGuire W, Black MM. Adult linear IgA disease and chronic bullous disease of childhood: the association with human lymphocyte antigens Cw7, B8, DR3 and tumour necrosis factor influences disease expression. Br J Dermatol. 1999 Nov;141(5):867-75. | PubMed |Patsatsi A. Chronic Bullous Disease or Linear IgA Dermatosis of Childhood –Revisited. J Genet Syndr Gene Ther. 2013;4:6 | CrossRef |Hirako Y, Usukura J, Uematsu J, Hashimoto T, Kitajima Y, Owaribe K. Cleavage of BP180, a 180-kDa bullous pemphigoid antigen, yields a 120-kDa collagenous extracellular polypeptide. J Biol Chem. 1998 Apr 17;273(16):9711-7. | PubMed |Zone JJ, Taylor TB, Meyer LJ, Petersen MJ. The 97 kDa linear IgA bullous disease antigen is identical to a portion of the extracellular domain of the 180 kDa bullous pemphigoid antigen, BPAg2. J Invest Dermatol. 1998 Mar;110(3):207-10. | PubMed |Allen J, Wojnarowska F. Linear IgA disease: the IgA and IgG response to dermal antigens demonstrates a chiefly IgA response to LAD285 and a dermal 180-kDa protein. Br J Dermatol. 2003 Nov;149(5):1055-8. | PubMed |Hendrix JD, Mangum KL, Zone JJ, Gammon WR. Cutaneous IgA deposits in bullous diseases function as ligands to mediate adherence of activated neutrophils. J Invest Dermatol. 1990 May;94(5):667-72.
| PubMed |Zone JJ, Egan CA, Taylor TB, Meyer LJ. IgA autoimmune disorders: development of a passive transfer mouse model. J Investig Dermatol Symp Proc. 2004 Jan;9(1):47-51. | PubMed |Caproni M, Rolfo S, Bernacchi E, Bianchi B, Brazzini B, Fabbri P. The role of lymphocytes, granulocytes, mast cells and their related cytokines in lesional skin of linear IgA bullous dermatosis. Br J Dermatol. 1999 Jun;140(6):1072-8. | PubMed |Mintz EM, Morel KD. Clinical features, diagnosis, and pathogenesis of chronic bullous disease of childhood. Dermatol Clin. 2011 Jul;29(3):459-62, ix. | CrossRef | PubMed |Weinberg MA, Insler MS, Campen RB. Mucocutaneous features of autoimmune blistering diseases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997 Nov;84(5):517-34. | PubMed |Shimizu S, Natsuga K, Shinkuma S, Yasui C, Tsuchiya K, Shimizu H. Localized linear IgA/IgG bullous dermatosis. Acta Derm Venereol. 2010 Nov;90(6):621-4. | CrossRef | PubMed |Burge S, Wojnarowska F, Marsden A. Chronic bullous dermatosis of childhood persisting into adulthood. Pediatr Dermatol. 1988 Nov;5(4):246-9. | PubMed |Verma R, Vasudevan B, Pragasam V, Dabbas D. Linear IgA disease in an adult with unusual clinical features. Indian Dermatol Online J. 2013 Apr;4(2):115-8. | CrossRef | PubMed |Aultbrinker EA, Starr MB, Donnenfeld ED. Linear IgA disease. The ocular manifestations. Ophthalmology. 1988 Mar;95(3):340-3. | PubMed |Godfrey K, Wojnarowska F, Leonard J. Linear IgA disease of adults: association with lymphoproliferative malignancy and possible role of other triggering factors. Br J Dermatol. 1990 Oct;123(4):447-52. | PubMed |Girăo L, Fiadeiro T, Rodrigues JC. Burn-induced linear IgA dermatosis. J Eur Acad Dermatol Venereol. 2000 Nov;14(6):507-10. | PubMed |Vargas TJ, Fialho M, Santos LT, Rodrigues PA, Vargas AL, Sousa MA. Linear IgA dermatosis associated with ulcerative colitis: complete and sustained remission after total colectomy. An Bras Dermatol. 2013 Jul-Aug;88(4):600-3. | CrossRef | PubMed |Collier PM, Kelly SE, Wojnarowska F. Linear IgA disease and pregnancy. J Am Acad Dermatol. 1994 Mar;30(3):407-11. | PubMed |Camilleri M, Pace JL. Drug-induced linear immunoglobulin-A bullous dermatosis. Clin Dermatol. 1998 May-Jun;16(3):389-91. | PubMed |Guide SV, Marinkovich MP. Linear IgA bullous dermatosis. Clin Dermatol. 2001 Nov-Dec;19(6):719-27. | PubMed |Reyes-Baraona F, Andino R, Carrasco JE, Arriagada C, Guerrero S. [Linear IgA bullous dermatosis of childhood: case report]. Arch Argent Pediatr. 2014 Apr;112(2):e57-60. | CrossRef | PubMed |Wojnarowska F, Marsden RA, Bhogal B, Black MM. Chronic bullous disease of childhood, childhood cicatricial pemphigoid, and linear IgA disease of adults. A comparative study demonstrating clinical and immunopathologic overlap. J Am Acad Dermatol. 1988 Nov;19(5 Pt 1):792-805. | PubMed |Willsteed E, Bhogal BS, Black MM, McKee P, Wojnarowska F. Use of 1M NaCl split skin in the indirect immunofluorescence of the linear IgA bullous dermatoses. J Cutan Pathol. 1990 Jun;17(3):144-8. | PubMed |Kuechle MK, Stegemeir E, Maynard B, Gibson LE, Leiferman KM, Peters MS. Drug-induced linear IgA bullous dermatosis: report of six cases and review of the literature. J Am Acad Dermatol. 1994 Feb;30(2 Pt 1):187-92. | PubMed |Bhogal B, Wojnarowska F, Marsden RA, Das A, Black MM, McKee PH. Linear IgA bullous dermatosis of adults and children: an immunoelectron microscopic study. Br J Dermatol. 1987 Sep;117(3):289-96. | PubMed |Allen J, Wojnarowska F. Linear IgA disease: the IgA and IgG response to the epidermal antigens demonstrates that intermolecular epitope spreading is associated with IgA rather than IgG antibodies, and is more common in adults. Br J Dermatol. 2003 Nov;149(5):977-85. | PubMed |Mintz EM, Morel KD. Treatment of chronic bullous disease of childhood. Dermatol Clin. 2011 Oct;29(4):699-700. | CrossRef | PubMed |Wojnarowska F. Linear IgA dapsone responsive bullous dermatosis. J R Soc Med. 1980 May;73(5):371-3. | PubMed |McFadden JP, Leonard JN, Powles AV, Rutman AJ, Fry L. Sulphamethoxypyridazine for dermatitis herpetiformis, linear IgA disease and cicatricial pemphigoid. Br J Dermatol. 1989 Dec;121(6):759-62. | PubMed |Mervic L, Dragos V, Pavlović MD. Linear IgA bullous dermatosis of childhood: successful treatment with miocamycin and topical corticosteroid. Clin Exp Dermatol. 2009 Oct;34(7):e391-2. | CrossRef | PubMed |Kasperkiewicz M, Schmidt E. Current treatment of autoimmune blistering diseases. Curr Drug Discov Technol. 2009 Dec;6(4):270-80. | PubMed |Alajlan A, Al-Khawajah M, Al-Sheikh O, Al-Saif F, Al-Rasheed S, Al-Hoqail I, et al. Treatment of linear IgA bullous dermatosis of childhood with flucloxacillin. J Am Acad Dermatol. 2006 Apr;54(4):652-6. | PubMed |Chaffins ML, Collison D, Fivenson DP. Treatment of pemphigus and linear IgA dermatosis with nicotinamide and tetracycline: a review of 13 cases. J Am Acad Dermatol. 1993 Jun;28(6):998-1000.
| PubMed |Peoples D, Fivenson DP. Linear IgA bullous dermatosis: successful treatment with tetracycline and nicotinamide. J Am Acad Dermatol. 1992 Mar;26(3 Pt2):498-9. | PubMed |Yomada M, Komai A, Hashimato T. Sublamina densa-type linear IgA bullous dermatosis successfully treated with oral tetracycline and niacianamide. Br J Dermatol. 1999 Sep;141(3):608-9. | PubMed |Khan IU, Bhol KC, Ahmed AR. Linear IgA bullous dermatosis in a patient with chronic renal failure: response to intravenous immunoglobulin therapy. J Am Acad Dermatol. 1999 Mar;40(3):485-8.
| PubMed |Kroiss MM, Vogt T, Landthaler M, Stolz W. High-dose intravenous immune globulin is also effective in linear IgA disease. Br J Dermatol. 2000 Mar;142(3):582. Erratum in: Br J Dermatol 2000 Jun;142(6):1268. | PubMed |Letko E, Bhol K, Foster CS, Ahmed AR. Linear IgA bullous disease limited to the eye: a diagnostic dilemma: response to intravenous immunoglobulin therapy. Ophthalmology. 2000 Aug;107(8):1524-8. | PubMed |Lozinski A, Baum S, Sagi L, Volkov A, Trau H, Barzilai A. [Rituximab (Mabthera) for treatment of rare autoimmune bullous skin disorders]. Harefuah. 2012 Oct;151(10):562-5, 606. | PubMed |Talhari C, Althaus C, Megahed M. Ocular linear IgA disease resulting in blindness. Arch Dermatol. 2006 Jun;142(6):786-7. | PubMed |






 Estudios originales
Estudios originales