Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Palabras clave: Trisomy 18 Syndrome, Tetralogy of Fallot, Ultrasonography, Prenatal, Survival, Abnormal Karyotype
El síndrome de Edwards o trisomía 18 es una entidad compleja, con afectaciones en los sistemas musculoesquelético, craneofacial, cardiovascular y neurológico. Su genética es variada, presentándose tanto de manera completa como en mosaicismo. Es infrecuente que la supervivencia supere el primer ańo de vida. Su caracterización fenotípica no es patognomónica, por lo cual el cariotipo es fundamental para el diagnóstico prenatal por medio de amniocentesis y cordocentesis mediante técnica de hibridación fluorescente in situ. Se presenta el caso de una paciente de ocho ańos que ha sobrevivido con esta condición, a pesar de presentar tetralogía de Fallot acompańada de malformaciones cardíacas graves. El diagnóstico comenzó por ecografía de tamizaje prenatal a las 16 semanas y ecografía de detalle, con amniocentesis y cariotipo de líquido amniótico, con resultado 47 XX+18. Ha sido tratada por múltiples especialidades médicas, debido a complicaciones osteomusculares, articulares, neurológicas, metabólicas y cardiovasculares que han limitado su calidad de vida. El manejo de estos pacientes requiere un equipo médico multidisciplinario. La consejería a los padres debe incluir aspectos relativos a la sobrevida, complicaciones frecuentes y riesgo-beneficio a evaluar antes de someter al menor a intervenciones quirúrgicas complejas o correctivas.
Ideas clave
|
El síndrome de trisomía 18 o síndrome de Edwards es polimalformativo, producido por la existencia de tres cromosomas 18. El genotipo completo se presenta en el 95% de los casos, ya sea por no disyunción o por translocación, aunque también se describe un genotipo incompleto en el 5% de los casos, pudiendo ser en mosaico o de trisomía parcial[1]. La trisomía 18 es la segunda cromosomopatía autosómica más frecuente después del síndrome Down o trisomía 21. La prevalencia global estimada es de uno por cada 6000 a 8000 recién nacidos y su frecuencia se incrementa en madres de edad avanzada[2].
Los pacientes con trisomía 18 tienen una elevada mortalidad desde el momento del nacimiento y a medida que avanza el tiempo menos son las probabilidades de sobrevivir, principalmente debido a su cardiopatía congénita. Sólo del 5 al 10% de los pacientes superan el primer año de vida. Se han reportado pocos casos de supervivencia en la literatura después de los cinco años[3], ya que se ha evidenciado que, de los que logran sobrevivir por encima del año de vida, sólo hasta 12% presentan supervivencia a esta edad[2],[3].
El diagnóstico de trisomía 18 se puede realizar a nivel prenatal por medio de ecografía de detalle, detectando malformaciones anatómicas. También es posible efectuarlo mediante pruebas de amniocentesis o cordocentesis, para realización de cariotipo o hibridación fluorescente in situ (del inglés Fluorescence in situ Hybridization, FISH)[4]. Después del nacimiento se diagnostica por medio de cariotipo bandas G en sangre periférica[5],[6]. El diagnóstico temprano incluye también determinaciones hormonales, previo a la realización de estudios invasivos[7].
Se presenta el caso de una paciente diagnosticada con trisomía 18 con una sobrevida mayor a los cinco años, poco frecuente con respecto a la reportado en la literatura, además de la presencia conjunta de cardiopatía congénita severa y múltiples manifestaciones del síndrome típicas y atípicas. El objetivo es aportar información acerca del conocimiento de esta trisomía en cuanto a características fenotípicas y sobrevida de los pacientes, lo cual es relevante en el campo de la genética, aspecto importante para establecer acciones diagnóstico-terapéuticas correctas.
Paciente de sexo femenino de ocho años, procedente de Pereira, Colombia. Es hija de padres no consanguíneos, con edades de 36 años para la madre y 35 años para el padre en el momento de la gestación. La paciente es producto del sexto embarazo, ya que las dos primeras gestas terminaron en aborto espontáneo y de la tercera a la quinta fueron de curso normal no presentándose cromosomopatías. La madre asistió a un total de siete controles prenatales, tuvo parto a término a las 39 semanas, no recibió orientación adicional sobre salud sexual y reproductiva ni tampoco consulta preconcepcional.
Durante los años previos a la consulta, la paciente recibió manejo interdisciplinario por pediatría, nutrición, cardiología, ortopedia y genética, con evolución limitada en su desarrollo. Los datos clínicos del presente reporte se recolectaron retrospectivamente a partir de una consulta de seguimiento y control por un nuevo pediatra, debido a su patología de base (trisomía 18), en el año 2020.
Al examen físico presentó las siguientes medidas antropométricas: talla 99 centímetros, peso 12 kilogramos, índice de masa corporal 12,2 kilogramos por metro cuadrado, con diagnóstico de desnutrición proteico-calórica severa crónica debido a que a lo largo de su vida se ha mantenido en percentiles inferiores para la edad, a pesar de tratamientos previos con suplementos alimenticios.
Con respecto a los hallazgos fenotípicos se encontró hipertelorismo, plagiocefalia, micrognatia con pabellones auriculares de implantación baja (Figura 1), fisura palatina y arco palatino estrecho (Figura 2), cejas pobladas con sinofris, hipertricosis (Figura 3), hipoplasia de las fosas nasales, cuello corto, escoliosis, luxación congénita de cadera bilateral con limitación para la abducción y flexión mayor a 90 grados, pie equinovaro izquierdo y pie talo-valgo derecho con atrofia muscular marcada (Figura 4), manos con desviación ulnar con segundo y quinto dedos superpuestos al tercero y cuarto e hipocratismo digital (Figura 5), retracciones en rodillas y tobillos, hipertricosis, hipotonía y cianosis central. A la auscultación cardíaca se encontró un soplo mesosistólico 4/6 en todos los focos auscultatorios.
Figura 1. Fotografía de la cabeza.
Figura 2. Fotografía de cavidad bucal.
Figura 3. Fotografía de rostro.
Figura 4. Fotografía de miembros inferiores.
Figura 5. Fotografía de ambas manos.
En ecografía de tamizaje realizada en la semana 12, se encontró feto con apariencia quística, septada a nivel de la nuca, edema de pie generalizado, derrame pericárdico, probable defecto del tabique interventricular a nivel de la cruz del corazón. En ecografía Doppler tricuspídea sedetectó regurgitación y en el de ductus venoso, flujo reverso. Además, se encontró translucencia nucal aumentada. Se solicitó biopsia de vellosidades coriales antes de la semana 14 o amniocentesis desde la semana 16. Se le realizó cariotipo de líquido amniótico y fetal mediante técnica de hibridación fluorescente in situ, con resultado 47 XX+18 en todas las mitosis analizadas, clínicamente compatible con trisomía 18.
Se le efectuó ecografía de detalle donde llamó la atención huesos tubulares entre 3 y 4 semanas, menores para la edad gestacional y un peso fetal estimado por debajo del percentil 3. A nivel cardíaco se observó defecto septal en el espacio intermembranoso de 2,8 milímetros asociado a cabalgamiento de la aorta, tracto de salida de ventrículo derecho aparentemente normal. En extremidades se observó angulación de pie respecto a las piernas. Adicionalmente, se detectó megacisterna magna y polihidramnios.
Al nacimiento presentó cuadro fenotípico y malformaciones compatibles con el trisomía 18. Debido a que la paciente ha tenido una sobrevida mayor a los tres meses, se hizo necesario establecer si la trisomía era libre o en mosaicismo. Para esto, se solicitó estudio citogenético con bandeo R en sangre periférica para definir cariotipo con resultado 47 XX+18 en un recuento de 20 metafases, esto es, una trisomía 18 libre. Se realizó el análisis citogenético a los padres y se encontró un mosaicismo en la madre 46 XX/47 XXX.
Desde el nacimiento se identificó tetralogía de Fallot, por lo cual permaneció con niveles de saturación de oxígeno no superiores al 80%. En el ecocardiograma se detectó comunicación interventricular e interauricular, aorta cabalgada al 45%, foramen oval permeable reverso, estenosis valvular pulmonar severa con gradiente máximo de 77 milímetros de mercurio y tronco de la arteria pulmonar dilatado. Con cierre de ductus arterioso después del tercer mes de vida, durante el primer año tuvo cinco seguimientos por cardiología pediátrica. La paciente usó en los tres primeros meses oxígeno domiciliario durante las 24 horas del día. Al segundo año de vida, se observaba cardiomegalia en la radiografía de tórax (Figura 6).
Figura 6. Radiografía de tórax.
A sus siete meses de vida, se describió equinovaro bilateral, deformidad en cavo asociado a enfermedad de tejidos blandos. Se le efectuaron imágenes de pelvis como se observa en la Figura 7. A los 11 meses de edad se le indicó yeso inquinopédico con rodilla en flexión de 80 grados, posterior a lo cual se le encontró mejoría del aducto de antepie de 20 grados. A los 17 meses se le indicó férula de Dennis Brown. A los cinco años, se identificó progresión con movilidad de las caderas con flexión hasta 90 grados y abducción de 10 grados con cifosis en unión toracolumbar. A raíz de ello, se concluye que la paciente posee luxación paralítica de sus caderas, escoliosis y deformidad en ambos pies que no requieren intervención quirúrgica.
Figura 7. Radiografía de pelvis.
Se le documentó hipotonía central severa desde su nacimiento, y retraso psicomotor secundarios a hipoxia crónica por cardiopatía compleja desde los primeros meses de vida. Por ecografía transfontanelar a los tres meses de edad, se observó verticalización de los cuernos frontales y probables signos de disgenesia del cuerpo calloso, sin otras alteraciones de la línea media ni compromiso hemorrágico. El estudio por resonancia magnética cerebral a los dos años reportó contenido intracraneal normal. Sin embargo, se identificaron cambios propios de hipoxia crónica, atrofia cortical cerebral marcada a nivel frontotemporal bilateral y retraso en la mielinización esperado para la edad.
Al momento de la elaboración de este reporte, la paciente no pronuncia sílabas, solo emite sonidos y su respuesta social es a través de la sonrisa, sostiene la cabeza, no realiza movimientos de desplazamiento. Al examen físico se evidenció hipoacusia izquierda, siendo capaz de comprender órdenes sencillas, sin control de esfínteres.
A la fecha de realización de este trabajo, la paciente tiene ocho años de edad, no ha presentado cuadros de convulsiones y está con manejo médico debido a que se consideró que no era candidata a cirugía cardiovascular por su pronóstico de vida poco favorable. No se han documentado malformaciones renales o digestivas en estudios ecográficos.
Para la publicación del caso clínico se obtuvo consentimiento informado de los padres de la paciente.
La literatura reporta que la mayoría de los fetos con trisomía 18 evolucionan a óbito. Si sobreviven hasta el nacimiento, la mayoría moriría en el primer año de vida[8]. Las niñas portadoras tendrían mayor probabilidad de supervivencia por periodos más largos, principalmente aquellas con defecto cromosómico por mosaicismo[9]. Aunque hay evidencia de la supervivencia a largo plazo, no se encuentra documentada en estudios poblacionales sino en reportes de caso. Aun así, se debe tener en cuenta que estos pacientes tienen un alto grado de dependencia y un retraso psicomotor severo[10],[11],[12],[13].
Se ha reportado una probabilidad mayor de mortalidad durante la primera semana de vida, siendo más alta en algunos países como Israel y Argentina[14],[15]. La mayoría de estos niños nacieron prematuros y la mayor supervivencia se logró hasta 39 días con una mediana de dos días[14],[15].
En un estudio poblacional en Estados Unidos, se encontró que la supervivencia a los cinco años fue del 12%, y los que sobrevivieron al primer año de vida tuvieron mayor probabilidad de sobrevivir hasta los cinco años. Las condiciones relacionadas con mayor supervivencia fueron sexo femenino, nacidos a término e hijos de madres negras no hispanas[16]. En otro estudio donde se encuestó a un grupo de neonatólogos con respecto al diagnóstico prenatal de trisomía 18, la mayoría estuvo de acuerdo con una supervivencia mínima de una semana y máxima de un año, donde el mejor pronóstico a nivel del neurodesarrollo sería un retraso mental profundo, y que sólo ofrecerían cuidado paliativo[17],[18].
Las principales complicaciones que generan mortalidad son las cardiopatías, apnea central e insuficiencia respiratoria[19],[20]. Por este motivo, llama la atención la sobrevida de esta paciente de ocho años, puesto que presenta una cardiopatía congénita severa la cual es infrecuente en los pacientes con el trisomía 18, además de no ser intervenida quirúrgicamente y teniendo en cuenta que menos del 4% de los casos sobreviven después del año de vida[2],[9]. El manejo de estos pacientes a lo largo de su vida incluye medidas invasivas como la reanimación cardiopulmonar por sus condiciones preexistentes, la nutrición enteral, uso de oxígeno y cirugías correctivas de defectos neurológicos o gastrointestinales principalmente[21].
Los defectos fenotípicos principalmente descritos son occipucio prominente, diámetro bifrontal estrecho, orejas de implantación baja, fisuras palpebrales, abertura oral y arco palatino estrechos, micrognatia e hirsutismo; de los cuales esta paciente presentó la mayoría de ellos. La plagiocefalia y la hipoplasia de las fosas nasales identificadas en la paciente, no están descritas claramente en la literatura. De los hallazgos a nivel del cráneo se describen, entre otros, hipertelorismo y paladar hendido; ambos identificados en la paciente[2],[9].
En el sistema cardiovascular hay manifestaciones frecuentes como el ductus arterioso persistente y defectos de los septos auriculares y ventriculares. En cuanto a manifestaciones medianamente frecuentes hay defectos de las valvas aórticas y pulmonares o estenosis pulmonar. En las menos frecuentes hay anomalías de las arterias coronarias, tetralogía de Fallot, dextrocardia y coartación de aorta, presentándose en la niña el diagnóstico de tetralogía de Fallot desde el nacimiento, condición que está presente en menos del 10%[2],[9].
A nivel abdomino-pélvico las malformaciones incluyen hernias umbilicales o inguinales, onfalocele, divertículo de Meckel, atresia esofágica, ano imperforado, riñón ectópico, doble uréter, tumor de Wilms o riñón poliquístico, condiciones que no presentó la menor[2],[9].
Se ha reportado que el 50% muestran deformidades posicionales del pie. La escoliosis es común en niños mayores y puede progresar entre los cinco y los 10 años. En la pelvis se ha descrito la luxación congénita de cadera que representa una condición poco frecuente y está presente en este caso. Otras malformaciones reportadas incluyen vértebras fusionadas, pectus excavatum, sindactilia de tercer y cuarto dedo de las manos y pies, cuello corto, escoliosis, desviación ulnar o radial y pie equinovaro. El caso reportado presentó las cuatro últimas, además de hipocratismo digital no mencionado en literatura[2],[9].
Los pacientes con trisomía 18 tienen más probabilidades de sobrevivir si se someten a cirugía cardíaca[21],[22]. En el estudio de Cooper y colaboradores, se evidenció que la cirugía cardíaca se asoció con una mejor supervivencia en pacientes con trisomía 18 (Odds ratio: 0,2; p = 0,02)[23]; sin embargo, el tratamiento debe ser individualizado a cada paciente[24]. En esta paciente no fue recomendable la realización de ningún procedimiento cardíaco ni cirugía debido al mal pronóstico, tomando en cuenta tanto el riesgo-beneficio, como los aspectos éticos y el consentimiento informado. Por otra parte, la oxigenoterapia para el manejo sintomático de la insuficiencia cardíaca fue suministrada a la paciente sólo por los primeros meses de vida.
Los pacientes con trisomía 18 presentan convulsiones en más del 50% de los casos, usualmente en el primer año de vida[25],[26],[27],[28]. Neurológicamente, se describen además mielinización escasa, hidrocefalia, defectos de cuerpo calloso y megacisterna magna. Los hallazgos presentados en este caso fueron estos dos últimos, además de la hipotonía, condición no reportada en la literatura[2],[9], la cual comúnmente describe hipertonía acompañando a este síndrome[6].
Aunque el genotipo más frecuente es la trisomía completa en todas las células, también existe la trisomía por mosaicismo y parcial. El fenotipo expresado después es el resultado de ese material genético adicional. Sin embargo, no existe un espectro definido patognomónico de trisomía 18, ya que las manifestaciones clínicas pueden ser diversas. Se ha postulado que existen unas regiones críticas en el cromosoma 18 para que se produzca como resultado un fenotipo. En los pacientes con mosaicismo, donde el cuadro clínico suele ser menos severo, las manifestaciones dependen de la proporción de células normales y afectadas. Aunque esto no se ha corroborado, ya que el fenotipo suele ser variable y es difícil establecer el porcentaje de células con trisomía 18. En algunos casos los padres no tenían ningún antecedente de cromosomopatía, es decir, esta es una entidad que puede transmitirse o ser de nueva aparición. Se debe tener en cuenta esta información al momento de realizar el consejo genético a los padres. Además, se debe explicar que la trisomía completa puede repetirse en un futuro embarazo con una probabilidad del 1%. En este caso el diagnóstico de la paciente es una trisomía 18 libre, aunque en el cariotipo efectuado se observaron únicamente veinte metafases y no cien, como es habitual[6],[11],[29],[30].
La frecuencia de anomalías macroscópicas detectadas ecográficamente en fetos con trisomía 18 varía con la edad gestacional, y su ocurrencia oscila entre un 53% hasta las 17,5 semanas de gestación y un 67% desde las 17,5 hasta las 24 semanas[31]. La afectación nucal, ya sea por el aumento de la translucencia (en el primer trimestre) o del pliegue (en el segundo) es el marcador ecográfico más confiable y ampliamente utilizado de las trisomías. En este caso clínico se identificó en ecografía temprana el aumento de la translucencia nucal. Con relación al aspecto quístico del feto, la alteración más frecuente son los higromas encontrados en el primer trimestre, los cuales se asocian con una trisomía, mientras que en el segundo trimestre a menudo se relaciona con la monosomía X[31],[32],[33]. Otro marcador frecuente en las trisomías son las alteraciones en el hueso nasal[31], en la ecografía de primer trimestre de la paciente se refirió a esta estructura como normal. Se ha encontrado que la ausencia de hueso nasal en trisomía 18 oscila entre 53 y 60%[34].
En relación con las características del Doppler, se identificaron el flujo venoso reverso y la regurgitación tricuspídea. La frecuencia de estos defectos es de 58 y 33,3% en pacientes con trisomía 18, respectivamente[34],[35],[36]. En ecografía de detalle del segundo trimestre, la paciente presentó un peso fetal estimado por debajo del percentil 3. Estudios han detectado aneuploidía en 20% de los fetos pequeños para edad gestacional[37],[38].
Otras alteraciones identificadas en ecografía incluyen quiste del plexo coroideo, defectos del tubo neural, ventriculomegalia, cráneo en forma de fresa, defectos multisistémicos (faciales, cardiovasculares, gastrointestinales, urogenitales), cordón umbilical de dos vasos, anormalidades de las extremidades, cisterna magna anormal, cuerpo calloso ausente o hipoplasia cerebelosa y polihidramnios[32]. De las anteriores, la paciente presentó estas cuatro últimas en el tamizaje prenatal.
Por último, cabe resaltar que la niña vive con ambos padres y estos han sido sus cuidadores permanentes desde su nacimiento, ya que la paciente es completamente dependiente para las actividades básicas de la vida diaria y se ha evidenciado que existe una adecuada red de apoyo familiar.
Se reporta el caso de una paciente femenina de 8 años con diagnóstico de trisomía 18 y cardiopatía congénita severa, teniendo en cuenta que son pocos los reportes donde se evidencia sobrevida mayor a 5 años, especialmente si se asocia con malformaciones cardiacas que son severas e infrecuentes.
Además, presentó manifestaciones no descritas en la literatura como la plagiocefalia, hipoplasia de fosas nasales, hipocratismo e hipotonía. Considerando que esta condición es infrecuente y de presentación atípica, se hace necesaria su descripción. El abordaje integral de estos pacientes requiere una intervención con varias especialidades médicas y debe realizarse una asesoría y consejería para los padres de manera individualizada, teniendo en consideración el pronóstico y las complicaciones del paciente.
Notas
Roles de contribución
VLR - EGM: conceptualización, metodología, investigación, preparación del manuscrito. VGZ - FAB: metodología, investigación, revisión y edición del manuscrito, visualización.
Conflictos de intereses
Los autores completaron la declaración de conflictos de interés de ICMJE y declararon que no recibieron fondos por la realización de este artículo; no tienen relaciones financieras con organizaciones que puedan tener interés en el artículo publicado en los últimos tres años y no tienen otras relaciones o actividades que puedan influenciar en la publicación del artículo. Los formularios se pueden solicitar contactando al autor responsable o al Comité Editorial de la Revista.
Financiamiento
Los autores no recibieron ningún tipo de financiamiento en este trabajo.
Consideraciones éticas
El consentimiento informado solicitado por Medwave para la publicación de este reporte de caso, incluyendo las imágenes mostradas, ha sido firmado por el tutor de la paciente. Los autores declaran que se respetó la privacidad de la paciente según las normas de CIOMS, de privacidad de los datos recolectados. Una copia del consentimiento informado fue remitida a la dirección editorial de la revista.
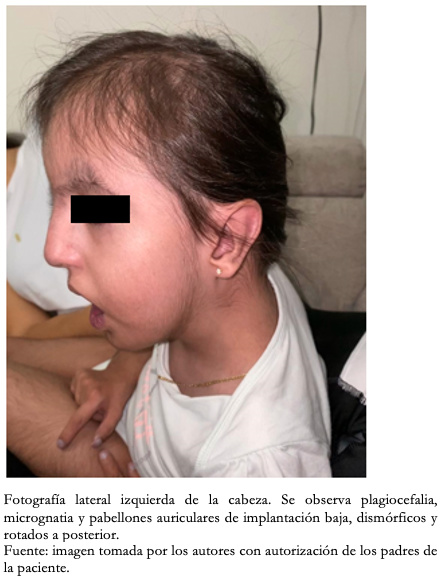 Figura 1. Fotografía de la cabeza.
Figura 1. Fotografía de la cabeza.
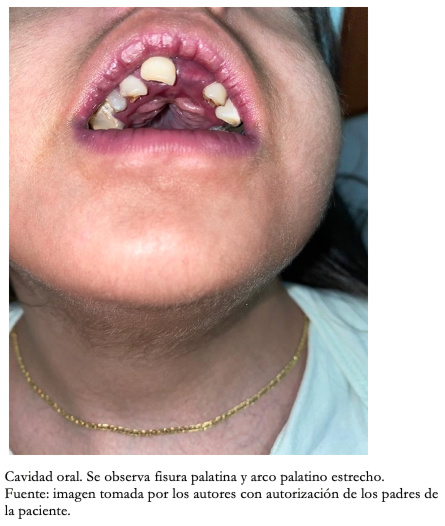 Figura 2. Fotografía de cavidad bucal.
Figura 2. Fotografía de cavidad bucal.
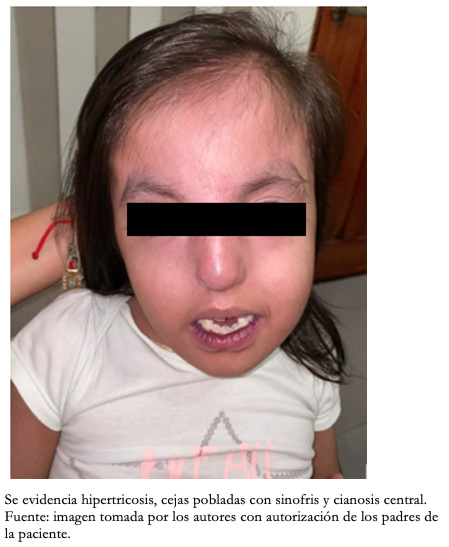 Figura 3. Fotografía de rostro.
Figura 3. Fotografía de rostro.
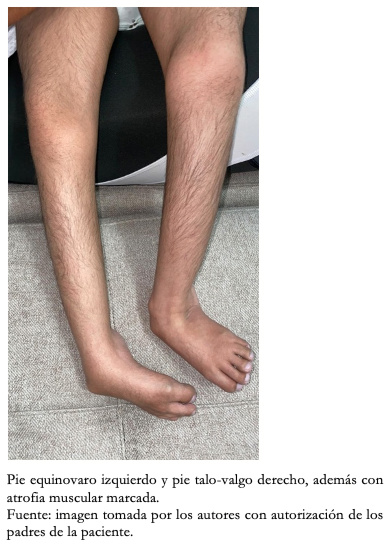 Figura 4. Fotografía de miembros inferiores.
Figura 4. Fotografía de miembros inferiores.
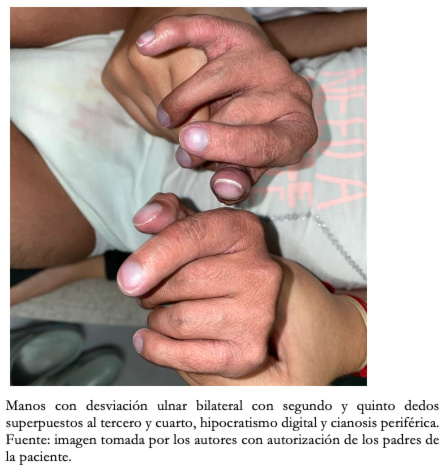 Figura 5. Fotografía de ambas manos.
Figura 5. Fotografía de ambas manos.
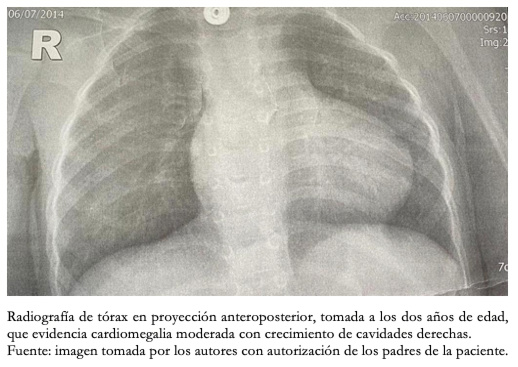 Figura 6. Radiografía de tórax.
Figura 6. Radiografía de tórax.
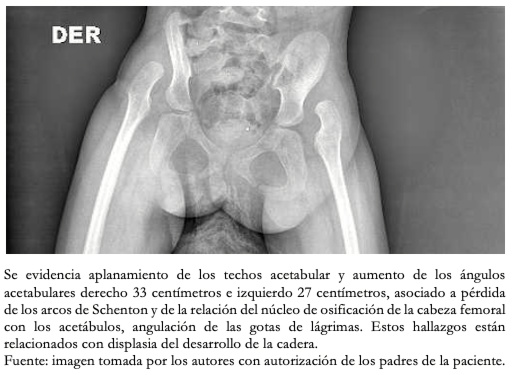 Figura 7. Radiografía de pelvis.
Figura 7. Radiografía de pelvis.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Edwards syndrome or trisomy 18 is a complex entity that involves the musculoskeletal, craniofacial, cardiovascular, and neurological systems. Its genetics are varied, presenting both in a complete and mosaic type. Survival rarely exceeds the first year of life. Its phenotype characterization is not pathognomonic, so karyotype is essential for diagnosis, prenatally by amniocentesis and cordocentesis by FISH technique. We present the case of an eight-year-old girl who has survived with this condition despite presenting tetralogy of Fallot and serious cardiac malformations. Diagnosis began with prenatal screening ultrasound at 16 weeks and detailed ultrasound, with amniocentesis and amniotic fluid karyotype, with a result of 47 XX+18. She has been treated by multiple medical specialties, due to musculoskeletal, joint, neurological, metabolic, and cardiovascular complications that have limited her quality of life. The management of these patients requires a multidisciplinary medical team, and counseling for parents should include aspects related to survival, frequent complications, and risk-benefit to be evaluated before subjecting the minor to complex or corrective surgical interventions.
Autores:
Valery López-Ríos[1], Estefanía Grajales-Marín[1], Valentina Gómez-Zambrano[1], Freddy Andrés Barrios-Arroyave[1]
Citación: López-Ríos V, Grajales-Marín E, Gómez-Zambrano V, Barrios-Arroyave FA. Prolonged survival in Edwards syndrome with congenital heart disease: a case report and literature review. Medwave 2020;20(8):e8015 doi: 10.5867/medwave.2020.08.8015
Fecha de envío: 12/6/2020
Fecha de aceptación: 15/8/2020
Fecha de publicación: 3/9/2020
Origen: No solicitado.
Tipo de revisión: Con revisión por pares externa, por tres árbitros a doble ciego.
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
del Gaudio L, Striano S, Coppola A. Severe epilepsy in an adult with partial trisomy 18q. Am J Med Genet A. 2014 Dec;164A(12):3148-53. | CrossRef | PubMed |Cereda A, Carey JC. The trisomy 18 syndrome. Orphanet J Rare Dis. 2012 Oct 23;7:81. | CrossRef | PubMed |Carey JC. Trisomy 18 and trisomy 13 syndromes. En: Cassidy SB, Allanson JE, editores. Management of genetic syndromes. 3rd ed. New York: John Wiley & Sons; 2010. p. 807-23. | CrossRef |Swedish Council on Health Technology Assessment. Methods of Early Prenatal Diagnosis: A Systematic Review (Summary and conclusions). Stockholm: Swedish Council on Health Technology Assessment (SBU); 2007. SBU Yellow Report No. 182. [Internet] | Link |Irving C, Richmond S, Wren C, Longster C, Embleton ND. Changes in fetal prevalence and outcome for trisomies 13 and 18: a population-based study over 23 years. J Matern Fetal Neonatal Med. 2011 Jan;24(1):137-41. | CrossRef | PubMed |Saldarriaga W, García-Perdomo HA, Arango-Pineda J, Fonseca J. Karyotype versus genomic hybridization for the prenatal diagnosis of chromosomal abnormalities: a metaanalysis. Am J Obstet Gynecol. 2015 Mar;212(3):330.e1-10. | CrossRef | PubMed |Huamán-Guerrero M, Quiroga DMM, Arias J, Huamán JM. Diagnóstico prenatal de anomalías cromosómicas. Biopsia de vellosidades coriales y amniocentesis para cariotipo fetal. Rev Per Ginecol Obstet 2007;53(3): 181-186. [Internet] | Link |Torres Hinojal MC, Marugán de Miguelsanz JM, Rodríguez Fernández LM. Supervivencia de 14 ańos en un paciente con síndrome de Edwards. An Pediatr (Barc) 2005;63(5):457-68. | CrossRef |Rosa RF, Rosa RC, Zen PR, Graziadio C, Paskulin GA. Trisomy 18: review of the clinical, etiologic, prognostic, and ethical aspects. Rev Paul Pediatr. 2013 Jan-Mar;31(1):111-20. | CrossRef | PubMed |Shanske AL. Trisomy 18 in a second 20-year-old woman. Am J Med Genet A. 2006 May 1;140(9):966-7. | CrossRef | PubMed |Saldarriaga W, Rengifo-Miranda H, Ramírez-Cheyne J. Síndrome de trisomía 18. Reporte de un caso clínico. Rev Chil Pediatr. 2016;87(2):129-136. | CrossRef |Ribeiro RR, dos Santos BM, Stuani AS, de Freitas AC, de Queiroz AM. Dental findings and dental care management in trisomy 18: case report of a 13-year-old "long-term survivor". Spec Care Dentist. 2006 Nov-Dec;26(6):247-51. | CrossRef | PubMed |Taksande A. A Rare Case Report of Edwards’s syndrome (Trisomy 18). EC Paediatrics. 2017;5(1):03-05. [Internet] | Link |Goel N, Morris JK, Tucker D, de Walle HEK, Bakker MK, Kancherla V, et al. Trisomy 13 and 18-Prevalence and mortality-A multi-registry population based
analysis. Am J Med Genet A. 2019 Dec;179(12):2382-2392. | CrossRef | PubMed |Duque JAP, Ferreira CF, Zachia SA, Sanseverino MTV, Gus R, Magalhăes JAA. The
natural history of pregnancies with prenatal diagnosis of Trisomy 18 or Trisomy
13: Retrospective cases of a 23-year experience in a Brazilian public hospital.
Genet Mol Biol. 2019;42(1 suppl 1):286-296. | CrossRef | PubMed |Meyer RE, Liu G, Gilboa SM, Ethen MK, Aylsworth AS, Powell CM, et al. Survival of
children with trisomy 13 and trisomy 18: A multi-state population-based study.
Am J Med Genet A. 2016 Apr;170A(4):825-37. | CrossRef | PubMed |Jacobs AP, Subramaniam A, Tang Y, Philips JB 3rd, Biggio JR, Edwards RK, et al. Trisomy 18: A survey of opinions, attitudes, and practices of
neonatologists. Am J Med Genet A. 2016 Oct;170(10):2638-43. | CrossRef | PubMed |Rosa RF, Rosa RC, Lorenzen MB, de Oliveira CA, Graziadio C, Zen PR, et al. Trisomy 18: frequency, types, and prognosis of congenital heart defects in a
Brazilian cohort. Am J Med Genet A. 2012 Sep;158A(9):2358-61. | CrossRef | PubMed |Kosho T, Nakamura T, Kawame H, Baba A, Tamura M, Fukushima Y. Neonatal
management of trisomy 18: clinical details of 24 patients receiving intensive
treatment. Am J Med Genet A. 2006 May 1;140(9):937-44. | CrossRef | PubMed |Springett A, Wellesley D, Greenlees R, Loane M, Addor MC, Arriola L, et al. Congenital anomalies associated with
trisomy 18 or trisomy 13: A registry-based study in 16 European countries,
2000-2011. Am J Med Genet A. 2015 Dec;167A(12):3062-9. | CrossRef | PubMed |Niedrist D, Riegel M, Achermann J, Schinzel A. Survival with trisomy 18--data
from Switzerland. Am J Med Genet A. 2006 May 1;140(9):952-9. | CrossRef | PubMed |Lorenz JM, Hardart GE. Evolving medical and surgical management of infants
with trisomy 18. Curr Opin Pediatr. 2014 Apr;26(2):169-76. | CrossRef | PubMed |Cooper DS, Riggs KW, Zafar F, Jacobs JP, Hill KD, Pasquali SK, et al. Cardiac Surgery
in Patients With Trisomy 13 and 18: An Analysis of The Society of Thoracic
Surgeons Congenital Heart Surgery Database. J Am Heart Assoc. 2019 Jul
2;8(13):e012349. | CrossRef | PubMed |Nelson KE, Rosella LC, Mahant S, Guttmann A. Survival and Surgical
Interventions for Children With Trisomy 13 and 18. JAMA. 2016 Jul
26;316(4):420-8. | CrossRef | PubMed |Roberts W, Zurada A, Zurada-ZieliŃSka A, Gielecki J, Loukas M. Anatomy of
trisomy 18. Clin Anat. 2016 Jul;29(5):628-32. | CrossRef | PubMed |Kumada T, Maihara T, Higuchi Y, Nishida Y, Taniguchi Y, Fujii T. Epilepsy in
children with trisomy 18. Am J Med Genet A. 2013 Apr;161A(4):696-701. | CrossRef | PubMed |Verrotti A, Carelli A, di Genova L, Striano P. Epilepsy and chromosome 18
abnormalities: A review. Seizure. 2015 Nov;32:78-83. | CrossRef | PubMed |Matricardi S, Spalice A, Salpietro V, Di Rosa G, Balistreri MC, Grosso S, et al. Epilepsy in the
setting of full trisomy 18: A multicenter study on 18 affected children with and
without structural brain abnormalities. Am J Med Genet C Semin Med Genet. 2016
Sep;172(3):288-95. | CrossRef | PubMed |Cammarata-Scalisi F, Lacruz-Rengel MA, Araque D, Da Silva G, Avendańo A,
Callea M, et al. Trisomía 18 en
mosaico. Serie de casos [Mosaic trisomy 18. Series of cases]. Arch Argent
Pediatr. 2017 Jun 1;115(3):e183-e186. | CrossRef | PubMed |Ahmad N, Wheeler K, Stewart H, Campbell C. Hepatoblastoma in a mosaic
trisomy 18 child with hemihypertrophy. BMJ Case Rep. 2016 Jan
21;2016:bcr2015211380. | CrossRef | PubMed |Bahado-Singh RO, Choi SJ, Oz U, Mendilcioglu I, Rowther M, Persutte W. Early second-trimester individualized estimation of trisomy 18 risk by ultrasound. Obstet Gynecol 2003; 101:463. [Internet] | CrossRef | Link |Rink BD, Norton ME. Screening for fetal aneuploidy. Semin Perinatol. 2016
Feb;40(1):35-43. | CrossRef | PubMed |Agathokleous M, Chaveeva P, Poon LC, Kosinski P, Nicolaides KH. Meta-analysis
of second-trimester markers for trisomy 21. Ultrasound Obstet Gynecol. 2013
Mar;41(3):247-61. | CrossRef | PubMed |Nicolaides KH. Screening for fetal aneuploidies at 11 to 13 weeks. Prenat
Diagn. 2011 Jan;31(1):7-15. | CrossRef | PubMed |Borrell A, Martinez JM, Farre MT, Azulay M, Cararach V, Fortuny A. Reversed
end-diastolic flow in first-trimester umbilical artery: an ominous new sign for
fetal outcome. Am J Obstet Gynecol. 2001 Jul;185(1):204-7. | CrossRef | PubMed |Florjański J, Fuchs T, Zimmer M, Homola W, Pomorski M, Blok D. The role of
ductus venosus Doppler flow in the diagnosis of chromosomal abnormalities during
the first trimester of pregnancy. Adv Clin Exp Med. 2013 May-Jun;22(3):395-401. | PubMed |






 Estudios originales
Estudios originales