Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Palabras clave: burkitt lymphoma, ovarian neoplasms, proto-oncogene proteins c-myc
La afectación ovárica como debut de un linfoma de Burkitt sin enfermedad extraovárica detectable es anecdótica, por lo que habitualmente no se incluye como hipótesis diagnóstica tras el hallazgo de una tumoración ovárica. Su desconocimiento lleva a realizar un tratamiento equivocado que puede llegar a comprometer el deseo reproductivo de la paciente. Presentamos el caso de una paciente que presenta un linfoma de Burkitt con afectación ovárica como manifestación inicial. La paciente desarrolló una progresión sistemática excepcionalmente rápida. A propósito de este caso y de su inusual evolución, revisamos la literatura existente.
La inclusión del ovario como manifestación inicial de un linfoma tipo Burkitt sin enfermedad extraovárica detectable es anecdótica. Es que el linfoma primario de ovario representa entre el uno y el 1,5% de todos los tumores ováricos. De estos, el linfoma de Burkitt primario de ovario representa tan solo 19% de los linfomas anexiales.
Debido a su infrecuencia, es por lo que habitualmente no se incluye como hipótesis diagnóstica tras el hallazgo de una tumoración ovárica, y el diagnóstico definitivo se alcanza con el estudio anatomopatológico. Este diagnóstico anatomopatológico se puede llevar a cabo atendiendo a particularidades inmunohistoquímicas del tumor, tales como la positividad para CD20 y CD10, así como para Ki-67 entre 90 y 100% de todos los núcleos celulares, junto con la demostración mediante prueba de hibridación in situ con fluorescencia (FISH) de una translocación del oncogén c-myc.
Alcanzar un adecuado diagnóstico resulta crucial, ya que se trata de un tumor quimiosensible y a pesar de que la cirugía es, en muchas ocasiones, necesaria en el diagnóstico y estadificación; esta no tiene que resultar una cirugía mutilante y, por lo tanto, no tiene que comprometer el deseo reproductivo de la mujer.
Presentamos el caso de una paciente de 13 años, la cual debuta con un abdomen agudo. Ante el hallazgo de una tumoración ovárica, precisó ser intervenida de urgencia. Posteriormente, el diagnóstico anatomopatológico informó de la presencia de un linfoma de Burkitt primario de ovario. Y a propósito del caso, se revisó la bibliografía existente.
Paciente de 13 años que es trasladada a urgencias hospitalarias por un cuadro de dolor intenso en hipogastrio y fosa iliaca derecha, agravado por un episodio sincopal. Refirió el inicio del dolor hace siete días, definiendo este como un dolor intermitente y de intensidad leve, con predominio nocturno, pero que permite el descanso. Durante este tiempo no presentó fiebre, presentó náuseas sin vómitos, así como tenesmo rectal, sin acusar otra clínica.
Durante la anamnesis no refirió antecedentes personales de interés. Como antecedentes ginecológicos destacó que todavía no ha presentado la menarquia y no ha iniciado relaciones sexuales.
A su ingreso, la paciente presentó intensa palidez, obnubilación e hipotensión marcada. Durante la auscultación cardíaca se objetivó una taquicardia con tonos apagados y la auscultación pulmonar resultó anodina. En la palpación abdominal se objetivó defensa involuntaria a la descomprensión brusca de cualquier región del abdomen y la auscultación abdominal puso de manifiesto una ausencia de peristalsis.
Tras la evaluación inicial, se realizó una ecografía abdominal en la que se determinó una masa sólida, alargada de 10 centímetros, localizada en hipogastrio junto con importante cantidad de líquido libre. De esta masa se extrajo una analítica que mostró una hemoglobina de 6,7 gramos por decilitros, hematocrito del 20% y 12 160 leucocitos con 70% de neutrófilos y 22% de linfocitos. La bioquímica informó de glucemia de 215 microgramos por decilitros, con función hepática y renal normal y proteína C reactiva de 0,31 microgramos por decilitros. La prueba de gestación aporta un resultado negativo.
Ante los hallazgos en la exploración física y las pruebas realizadas, se decidió laparotomía urgente en la que se encontró un importante hemoperitoneo de 2000 mililitros, además de abundantes coágulos y la presencia de una masa sólida alargada de 12 por 10 por 4 centímetros dependiente de anejo derecho y enclavada en Douglas, la cual presentó una superficie externa lobulada y rugosa de color beige tostado con estriaciones a través de las cuales sangra de forma profusa (Figura 1). El resto de la exploración de cavidad pélvica y abdominal resultó anodina con útero y ovario izquierdo normal. Tras los hallazgos citados se procedió a anexectomía derecha y lavado de cavidad y se remitió muestra a anatomía patológica, donde en una primera inspección de la pieza se describió una sustitución difusa del ovario por tumoración de superficie lisa y brillante, homogénea y sonrosada de consistencia elástica (Figura 2).
Figura 1. Tumoración de 12 x 10 x 4 cm.
Figura 2. Detalle de apertura de la pieza.
Durante el postoperatorio inmediato, la paciente precisó de transfusión de sangre y plasma fresco congelado e insulinoterapia. Los días posteriores la evolución de la paciente fue favorable, por lo que se procedió al alta hospitalaria en el cuarto día postquirúrgico con buen estado general, bajo profilaxis antibiótica. Quedó pendiente el resultado definitivo de anatomía patológica.
Veintiún días después de la cirugía, la paciente ingresó de nuevo por dolor en epigastrio, vómitos, astenia e intolerancia alimenticia. En la exploración física, llamó la atención un regular estado general y una intensa palidez cutánea y a nivel abdominal. Se palpó una tumoración subcutánea en zona umbilical, así como múltiples nódulos pequeños subcutáneos a lo largo de toda la incisión de laparotomía previa. La analítica en este nuevo ingreso mostró un hemograma normal (hemoglobina de 11 gramos por decilitro, hematocrito 35%, 14 470 leucocitos (57% de neutrófilos), plaquetas 172 000) aunque resultó remarcable los cambios hallados en la bioquímica, tales como una elevación de la GPT/ALT (823 unidades por litro), así como de la amilasa (237 unidades por litro), fosfatasa alcalina (1169 unidades internacionales por litro) y lactato deshidrogenasa (1400 unidades por litro). El resto de la bioquímica fue normal y la radiografía de tórax también resultó normal.
En la ecografía abdominal realizada durante la valoración ginecológica, se observó un hematoma organizado en diferentes estadios de evolución de 10 centímetros en pared abdominal y pequeños nódulos anecoicos a lo largo de toda la zona de incisión laparotómica, junto con ausencia de líquido libre intraperitoneal. A nivel pélvico se visualizó un ovario izquierdo aumentado de tamaño, aunque de ecoestructura normal y útero de morfología y tamaño normal. Sistemáticamente se realizó una revisión del resto de la cavidad abdominal, resultando llamativo la presencia de varios nódulos de tres a cuatro centímetros hipoecogénicos y de estructura interna desorganizada en cabeza, cuerpo y cola de páncreas (Figura 3).
Figura 3. Ecografía abdominal.
Ante la sospecha de que las imágenes visualizadas en la ecografía pudiesen corresponder con nódulos metastásicos de pared abdominal, incisión laparotómica y páncreas, se solicitó una resonancia magnética nuclear. Ésta informó una masa en cabeza de páncreas de 4,9 centímetros, junto con un aumento de volumen difuso del cuerpo pancreático y la cola del páncreas, así como de la presencia de adenopatías retroperitoneales y alguna adenopatía mesentérica. A nivel pélvico, se reconoció una tumoración irregular de gran volumen que afectaba a la pared abdominal y el ovario izquierdo se encontró aumentado de tamaño, aunque conteniendo folículos.
En esta encrucijada diagnóstica, los resultados definitivos de anatomía patológica de la pieza quirúrgica resultaron esclarecedores. Se trataba de un tumor compuesto por una proliferación difusa de células monomórficas de tamaño medio, con núcleos redondos, nucléolos múltiples y escaso citoplasma basófilo (Figura 4).
Figura 4. Proliferación difusa de células monomórficas.
El estudio inmunohistoquímico reveló que dichas células mostraron positividad para CD20, CD79a y CD10, lo que indicó que se trataba de una población de linfocitos de estirpe B (Figura 5). Intercalados entre estos linfocitos atípicos, se identificaron macrófagos marcados con CD68 que tratan de ingerir células apoptóticas, dando una imagen típica en cielo estrellado (Figura 6). La intensa proliferación celular quedó patente con la tinción en prácticamente el 100% de los núcleos celulares para Ki67 (Figura 7). También se objetivó que el tumor no expresa BCL2, BCL6, así como tampoco expresó marcadores de células de estirpe T como CD5 y CD3. El análisis de hibridación fluorescente in situ a partir de secciones de tejido del ovario, informó de una rotura de cMyc (8q24) en un 25% de los núcleos. La serología de VIH, VHA, VHC, así como los anticuerpos virales de Epstein-Barr, también resultaron negativos.
Figura 5. Tinción inmunohistoquímica positiva para CD20.
Figura 6. Patrón de cielo estrellado.
Figura 7. Tinción inmunohistoquímica positiva para Ki 67.
El análisis posterior de punción lumbar resultó positivo para células tumorales. La citometría de médula ósea reveló 280000 células por microlitro, con una población de linfocitos B de elevado tamaño y granulación, que representan el 37% del total de células, con fenotipo CD19+ CD10+ CD20+, CD34+ CD38+ CD79b+ Tdt y monoclonalidad Inmunoglobulinas /ƛ, con expresión parcialmente positiva de CD15, pero sin otros marcadores mieloides.
Ante las características morfológicas y el inmunoperfil citado, la paciente fue diagnosticada de linfoma de Burkitt primario de ovario, con rápida progresión sistémica.
Posteriormente, tras el adecuado diagnóstico, fue tratada con una combinación de agentes quimioterapéuticos entre los que se incluyeron doxorrubicina, vincristina, citarabina, genoxal y dexametasona, sin efectos secundarios importantes. Estos agentes se combinaron con anticuerpo anti-CD20 (Rituximab), así como metotrexate intratecal. Tras cuatro años desde el inicio del tratamiento, la paciente se encuentra libre de enfermedad y eumenorreica.
Desde que Dennis Burkitt describiese el primer caso de linfoma de Burkitt en 1958, mucho se ha investigado sobre él y gracias a ello conocemos que se trata de un linfoma altamente agresivo, aunque potencialmente curable, con una mayor prevalencia en mujeres que en varones y con una edad media de diez años en su debut[1],[2].
La Organización Mundial de la Salud (OMS) aporta una definición histológica de linfoma de Burkitt congruente con la presentada en el caso clínico, definiéndolo como un tumor compuesto por células B monomórficas de tamaño intermedio, con núcleos redondos, nucléolos múltiples y escaso citoplasma basófilo junto con numerosas figuras mitóticas atípicas[3]. En este tumor resulta característica la tasa excepcionalmente alta de proliferación y apoptosis celular, así como de muerte celular espontánea, lo que queda patente a través del típico “patrón en cielo estrellado” impartido por los macrófagos que ingieren las células tumorales apoptóticas.
Desde la perspectiva inmunohistoquímica, el linfoma de Burkitt presenta unas particularidades que ayudan a su diagnóstico. Una de estas es la expresión de la proteína c-myc, que resulta como consecuencia de la translocación en el cromosoma 8q24, otra de ellas en la positividad para el antígeno Ki67 en un orden del 90 al 100%. Ello se traduce en un alto índice de proliferación de la población tumoral y, por otro lado, la ausencia de la oncoproteína bcl-2[4],[5]. Estas características inmunohistoquímicas han sido apoyadas por autores como Haralambieva y sus colaboradores[6], quienes proponen los siguientes criterios diagnósticos para el linfoma de Burkitt, todos los cuales deben cumplirse: una positividad para ki-67 de al menos 90%, positividad para CD10, falta de expresión de Bcl-2 y la presencia de un punto de ruptura en la región Myc. Sin embargo, estos criterios han sido matizados en estudios posteriores como el publicado por la OMS, en el que se argumenta que hasta en el 10% de los casos de linfoma de Burkitt puede faltar el reordenamiento de c-Myc[1]. En el caso que presentamos, las características inmunohistoquímicas del tumor coinciden con las clásicamente propuestas por Haralambieva, lo que facilitó el llegar a un juicio diagnóstico de linfoma de Burkitt.
El debut primario en ovario es excepcional, resultando comprometida esta localización principalmente como parte de un proceso de enfermedad generalizada. El linfoma primario de ovario representa entre el uno y 1,5% de todos los tumores ováricos y de estos, el linfoma de Burkitt primario de ovario representa tan solo 19% de los linfomas anexiales. El diagnóstico pasa desapercibido ante la presencia de un tumor anexial en el contexto de un abdomen agudo debido a su infrecuencia, como es en nuestro caso[7].
A la rareza de esta entidad, se une la controversia que existe en torno al origen del linfoma primario de ovario. Monterroso y sus colaboradores intentaron argumentarlo al demostrar la presencia de tejido linfoide en ovarios normales, aunque en escasa cantidad[8]. Sin embargo, otros autores han defendido que el origen del linfoma primario de ovario estaría secundado por procesos inflamatorios que involucrarían al ovario tales como endometriosis, enfermedad inflamatoria pélvica o enfermedades autoinmunes[9],[10].
Independientemente de su origen, es extremadamente importante diferenciar entre la afectación primaria y secundaria del tumor, ya que la afectación primaria, pese a su comportamiento agresivo, muestra tasas de supervivencia del 80%a los cinco años, en comparación con las tasas de supervivencia en la afectación secundaria las cuales caen dramáticamente a un 33% en ese mismo período. Para realizar un adecuado diagnóstico, en 1976 Fox y Langley[11],[12] establecieron una serie de criterios, según los cuales un linfoma podría catalogarse como primario de ovario si el linfoma estaba confinado clínicamente al ovario (considerando confinamiento clínico a la afectación de los ganglios linfáticos adyacentes inmediatos o a estructuras adyacentes inmediatas), junto con la falta de evidencia del linfoma en ningún otro lugar en el momento del diagnóstico. Como segundo criterio, establecieron que debía haber un estudio inicial de sangre periférica y médula ósea negativo para células tumorales. Por último, dictaminaron que debían transcurrir varios meses entre el debut del tumor anexial y la aparición de lesiones linfomatosas adicionales en áreas alejadas del ovario.
Si atendemos a los criterios de 1976 de Fox y Langley, nuestro caso clínico debutó con la presencia de un linfoma confinado clínicamente al ovario tipo Burkitt, junto con la falta de evidencia del linfoma en ningún otro lugar en el momento de la cirugía inicial. Sin embargo, su rápida progresión y la aparición de lesiones linfomatosas adicionales macroscópicamente visibles en áreas alejadas del ovario en tan corto periodo de tiempo, hacen de éste, un caso excepcional.
Atendiendo a estos criterios diagnósticos y realizando una revisión de la literatura, solamente diecisiete casos de linfoma de Burkitt primario de ovario han sido documentados en la literatura hasta la fecha. En general, el prototipo de paciente y la evolución clínica se repite en todas estas pacientes.
El linfoma de Burkitt primario de ovario se suele presentar en mujeres jóvenes, con una edad media de 20 años a su diagnóstico. En los siete casos descritos en la literatura en los que se mencionan la duración de los síntomas al diagnóstico, los mismos acontecieron de forma brusca, con un episodio de dolor abdominal y distensión o presentaron una clínica insidiosa provocada por la compresión tumoral, en las dos a seis semanas previas, son coincidentes con el caso que presentamos.
Debido a este debut agudo, diez pacientes fueron sometidas a cirugía antes del diagnóstico y pese a que al 40% se les realizó una biopsia intraoperatoria, ninguna de las mismas concluyó con el diagnóstico de linfoma de Burkitt. En este punto existe una conexión en el hecho del desconocimiento del diagnóstico al debut del cuadro y la demora en el mismo hasta el resultado anatomopatológico. Por este motivo, el ser conocedores de esta entidad puede resultar crucial a la hora de plantear un adecuado diagnóstico diferencial. En cuanto al pronóstico de los casos reportados en la literatura, los resultados fueron dispares, aunque este dato podría encontrarse sesgado por el tratamiento recibido, ya que la mayoría de los casos se presentaron en las décadas de 1970 y 1980[13],[14],[15],[16],[17],[18],[19],[20],[21],[22],[23],[24],[25],[26].
En el caso que presentamos, la paciente respondió de manera satisfactoria al tratamiento con quimioterapia. Actualmente se encuentra libre de enfermedad presentando un pronóstico acorde con el del linfoma de Burkitt primario de ovario. Este dato apoya la hipótesis diagnóstica inicial.
La afectación ovárica como manifestación inicial de un linfoma de Burkitt resulta un hecho anecdótico y precisa de una combinación de hallazgos clínicos, inmunológicos y moleculares para realizar un correcto diagnóstico, y consecuentemente adoptar una adecuada conducta terapéutica que no resulte mutilante para la paciente.
Por lo tanto, los profesionales sanitarios deben ser conocedores de esta entidad con el fin de poder realizar un adecuado diagnóstico diferencial, en una tumoración anexial en una paciente en edad fértil.
Roles y contribuciones de autoría
AGA concibió y diseñó el estudio. MNQ y FPO participaron en el análisis de la literatura existente. AGA, FPO, MNQ, MDS, JQV y JFA participaron en la realización del artículo, la obtención de los datos clínicos y, la interpretación de los resultados de las pruebas complementarias así como en la redacción y la revisión crítica del manuscrito.
Financiamiento
Los autores del presente artículo declaran que no han recibido ningún tipo de financiación.
Conflictos de intereses
Los autores del presente artículo declaran que no existen conflictos de interés.
Aspectos éticos
El consentimiento informado solicitado por Medwave, ha sido firmado por el paciente; una copia de este fue remitido a la dirección editorial de la Revista
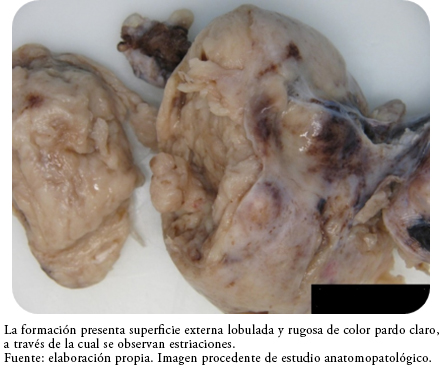 Figura 1. Tumoración de 12 x 10 x 4 cm.
Figura 1. Tumoración de 12 x 10 x 4 cm.
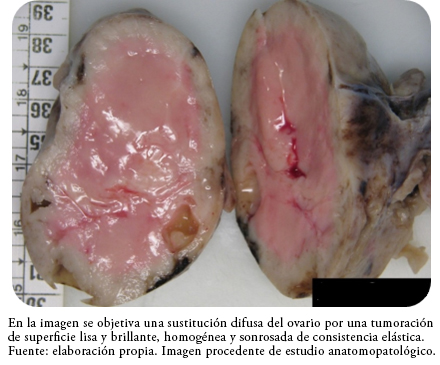 Figura 2. Detalle de apertura de la pieza.
Figura 2. Detalle de apertura de la pieza.
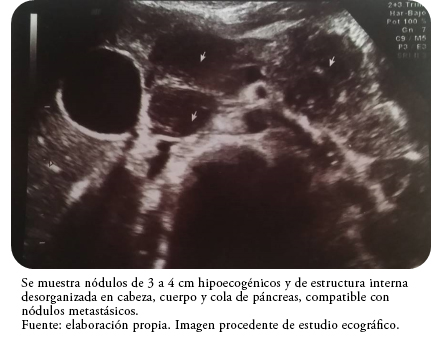 Figura 3. Ecografía abdominal.
Figura 3. Ecografía abdominal.
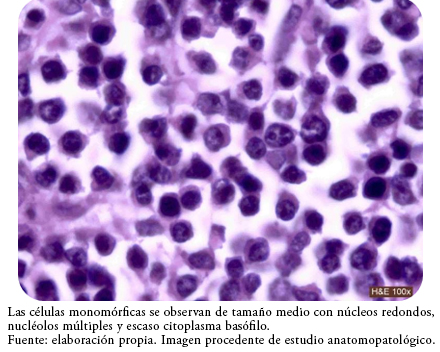 Figura 4. Proliferación difusa de células monomórficas.
Figura 4. Proliferación difusa de células monomórficas.
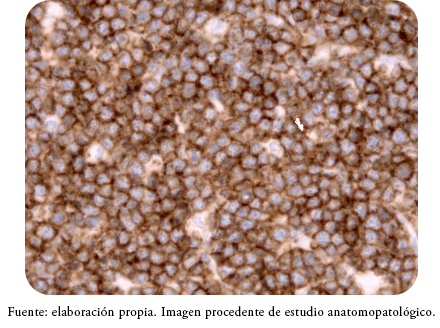 Figura 5. Tinción inmunohistoquímica positiva para CD20.
Figura 5. Tinción inmunohistoquímica positiva para CD20.
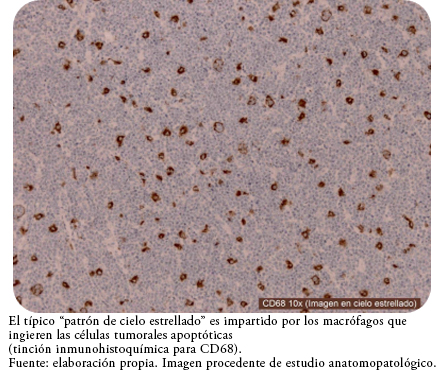 Figura 6. Patrón de cielo estrellado.
Figura 6. Patrón de cielo estrellado.
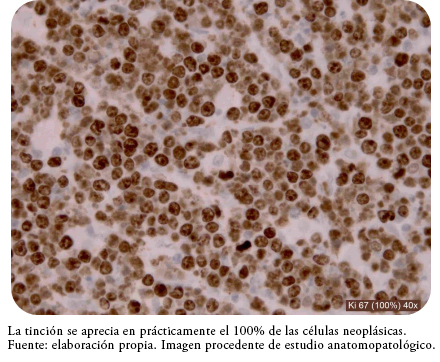 Figura 7. Tinción inmunohistoquímica positiva para Ki 67.
Figura 7. Tinción inmunohistoquímica positiva para Ki 67.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Ovarian involvement as the initial manifestation of a Burkitt lymphoma without detectable extra-ovarian disease is rare, which is why it is usually not included in the differential diagnosis when an ovarian tumor is detected. A missed diagnosis will lead to the wrong treatment being given, and this can compromise any future reproductive wishes of the patient. In this article, a patient presents a Burkitt lymphoma with ovarian involvement as an initial manifestation and an unusually rapid systemic progression of the disease. Prompted by this case and its unusual course, we reviewed the existing literature.
Autores:
Ana Gómez Alarcón[1], María Nieves Quesada Fernández[1], Fátima Parras Onrubia[1], María Dolores Díaz Serrano[2], José Quesada Villar[1], José Fernando Hernández Aznar[3]
Citación: Gómez Alarcón A, Quesada Fernández MN, Parras Onrubia F, Díaz Serrano MD, Quesada Villar J, Hernández Aznar JS. Ovarian Burkitt lymphoma as the primary manifestation: A case report and literature review. Medwave 2019;19(7):e7674 doi: 10.5867/medwave.2019.07.7674
Fecha de envío: 5/11/2018
Fecha de aceptación: 24/6/2019
Fecha de publicación: 2/8/2019
Origen: no solicitado
Tipo de revisión: Con revisión por pares externa por cuatro pares revisores, a doble ciego
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Jaffe ES, Harris NL, Stein H, Isaacson PG. Classification of lymphoid neoplasms: the microscope as a tool for disease discovery. Blood. 2008 Dec 1;112(12):4384-99. | CrossRef | PubMed |Ferry JA. Burkitt’s lymphoma: Clinicopathologic features and differential diagnosis. Oncologist 2006; 11: 375-383. | CrossRef |Swerdlow SH, Campo E, Harris NL. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (4th ed). Lyon, France: International Agency for Research on Cancer; 2008. | Link |Dalla-Favera R. Molecular genetics of aggressive B-cell lymphoma. Hematol Oncol. 2017 Jun;35 Suppl 1:76-79. | CrossRef | PubMed |Molyneux EM, Rochford R, Griffin B, Newton R, Jackson G, Menon G, et al. Burkitt's lymphoma. Lancet. 2012 Mar 31;379(9822):1234-44. | CrossRef | PubMed |Haralambieva E, Boerma EJ, van Imhoff GW, Rosati S, Schuuring E, Müller-Hermelink HK, et al. Clinical, immunophenotypic, and genetic analysis of adult lymphomas with morphologic features of Burkitt lymphoma. Am J Surg Pathol. 2005 Aug;29(8):1086-94. | PubMed |Rostion CG. Mesenteric adenitis and gynecological problems that cause acute abdominal pain. Medwave 2005 Ene;5(1):e1948. | CrossRef |Monterroso V, Jaffe ES, Merino MJ, Medeiros LJ. Malignant lymphomas involving the ovary. A clinicopathologic analysis of 39 cases. Am J Surg Pathol. 1993 Feb;17(2):154-70. | PubMed |Crawshaw J, Sohaib SA, Wotherspoon A, Shepherd JH. Primary non-Hodgkin's lymphoma of the ovaries: imaging findings. Br J Radiol. 2007 Aug;80(956):e155-8.
| CrossRef | PubMed |Chishima F, Hayakawa S, Ohta Y, Sugita K, Yamazaki T, Sugitani M, Yamamoto T: Ovarian Burkitt´s lymphoma diagnosed by a combination of clinical features, morphology, immunophenotype, and molecular findings and successfully managed with surgery and chemotherapy. J Gynecol Cancer 2006, 16(1):337-343. | CrossRef |Fox H, Langley FA (eds): Malignant lymphoma of ovary. In: Tumors of the ovary. London: Heinemann, 1976: 293-9. | Link |Fox H, Langley FA, Govan AD, Hill AS, Bennett MH. Malignant lymphoma presenting as an ovarian tumour: a clinicopathological analysis of 34 cases. Br J Obstet Gynaecol. 1988 Apr;95(4):386-90. | PubMed |Chorlton I, Norris HJ, King FM. Malignant reticuloendothelial disease involving the ovary as a primary manifestation: a series of 19 lymphomas and 1 granulocytic sarcoma. Cancer. 1974 Aug;34(2):397-407. | PubMed |Arseneau JC, Canellos GP, Banks PM, Berard CW, Gralnick HR, DeVita VT Jr: American Burkitt’s lymphoma: a clinicopathologic study of 30 cases. I. Clinical factors relating to prolonged survival. Am J Med 1975; 58: 314-321. | PubMed |Vang R, Medeiros LJ, Warnke RA, Higgins JP, Deavers MT: Ovarian non-Hodgkin’s lymphoma: a clinicopathologic study of eight primary cases. Mod Pathol 2001; 14: 1093-1099. | CrossRef |Baloglu H, Turken O, Tutuncu L, Kizilkaya E. 24-year-old female with amenorhea: bilateral primary ovarian Burkitt lymphoma. Gynecol Oncol. 2003 Nov;91(2):449-51. | PubMed |Chishima F, Hayakawa S, Ohta Y, Sugita K, Yamazaki T, Sugitani M, et al. Ovarian Burkitt's lymphoma diagnosed by a combination of clinical features, morphology, immunophenotype, and molecular findings and successfully managed with surgery and chemotherapy. Int J Gynecol Cancer. 2006 Jan-Feb;16 Suppl 1:337-43. | PubMed |Lu SC, Shen WL, Cheng YM, Chou CY, Kuo PL. Burkitt'S lymphoma mimicking a primary gynecologic tumor. Taiwan J Obstet Gynecol. 2006 Jun;45(2):162-6. | PubMed |Ng SP, Leong CF, Nurismah MI, Shahila T, Jamil MA. Primary Burkitt lymphoma of the ovary. Med J Malaysia. 2006 Aug;61(3):363-5. | PubMed |Crawshaw J, Sohaib SA, Wotherspoon A, Shepherd JH. Primary non-Hodgkin's lymphoma of the ovaries: imaging findings. Br J Radiol. 2007 Aug;80(956):e155-8. | PubMed |Muńoz Martín AJ, Pérez Fernández R, Vińuela Beneítez MC, Martínez Marín V, Marquéz-Rodas I, Sabin Domínguez P, et al. Primary ovarian Burkitt lymphoma. Clin Transl Oncol. 2008 Oct;10(10):673-5. | PubMed |Gutiérrez-García L, Medina Ramos N, García Rodríguez R, Barber MA, Arias MD, García JA. Bilateral ovarian Burkitt's lymphoma. Eur J Gynaecol Oncol. 2009;30(2):231-3. | PubMed |Cui T, Wang C, He H, Shi G, Hu L. A rare case of ovarian Burkitt lymphoma associated tumor lysis syndrome. Eur J Gynaecol Oncol. 2010;31(2):209-10. | PubMed |Cyriac S, Srinivas L, Mahajan V, Sundersingh S, Sagar TG. Primary Burkitt's lymphoma of the ovary. Afr J Paediatr Surg. 2010 May-Aug;7(2):120-1.
| CrossRef | PubMed |






 Estudios originales
Estudios originales