Revista Biomķdica Revisada Por Pares

 Para Descargar PDF debe Abrir sesi¾n.
Para Descargar PDF debe Abrir sesi¾n.
Palabras clave: multiple endocrine neoplasia type 2A, pheochromocytoma, abdominal pain, pulmonary tuberculosis
El feocromocitoma constituye una neoplasia productora de catecolaminas que se presenta de forma esporßdica o asociada a enfermedades de transmisi¾n hereditaria, como la neoplasia endocrina m·ltiple. Los sĒntomas clßsicos como la cefalea, sudoraci¾n y palpitaciones son atribuidos a la actividad del sistema nervioso simpßtico y suelen presentarse en forma de paroxismos. La tuberculosis pulmonar es una enfermedad infecciosa que constituye un problema de salud p·blica en muchos paĒses, cuya incidencia depende de algunos factores incluyendo la inmunosupresi¾n que generan las enfermedades endocrino-tumorales como la antes descrita. Presentamos el caso de un paciente masculino de 38 a±os que acude a emergencia por presentar un paroxismo de hipertensi¾n arterial y dolor abdominal, como manifestaciones iniciales de un feocromocitoma en el contexto de una neoplasia endocrina m·ltiple de tipo IIA. El paciente desarroll¾ de forma concomitante tuberculosis pulmonar; no obstante, se logr¾ tratar ambas entidades consiguiendo una evoluci¾n clĒnica favorable.
El feocromocitoma es un tumor productor de catecolaminas que, aunque raro, puede derivar en un resultado fatal para el portador si no es diagnosticado de forma oportuna. Las manifestaciones son diversas y el tumor puede imitar una variedad de condiciones, a menudo dando como resultado un diagnóstico erróneo y diferido[1]. Este puede presentarse de forma esporádica y en una proporción mucho menos frecuente, asociado a los cuadros de neoplasia endocrina múltiple como el caso que describimos[2]. La tríada clásica de síntomas incluye cefalea, sudoración y taquicardia[3].
Presentamos el caso de un paciente varón joven que, además de los síntomas mencionados, presentó dolor abdominal como manifestación clínica de dicha enfermedad. Se trata de una manifestación bastante inusual y poco descrita en esta condición, además de padecer una enfermedad endémica en Perú como es la tuberculosis.
Información del paciente
Varón de 38 años oriundo de la ciudad de Pucallpa, ubicada en la selva de Perú, de ocupación obrero de construcción, sin historia personal ni familiar de enfermedades ni hábitos nocivos. Acudió a emergencia hospitalaria en su localidad por presentar durante 48 horas dolor abdominal sordo en mesogastrio, irradiado en banda, intenso (escala análoga visual 7/10) y permanente. En el trascurso de las horas se agregó malestar general, sudoración profusa y cefalea súbita holocraneana intensa (9/10) asociado a mareos, visión borrosa e inestabilidad a la bipedestación.
Hallazgos clínicos
Al examen físico de ingreso se registró presión arterial de 200/130 milímetros de mercurio, sin taquicardia, sin taquipnea y pulsoximetría normal. A nivel abdominal, los ruidos hidroaéreos estaban presentes con timpanismo a la percusión. La palpación profunda del abdomen era dolorosa de forma difusa, sin reacción peritoneal. No se palparon masas. No presentó otros hallazgos clínicos relevantes en el resto de la exploración física.
Evaluación diagnóstica
Los valores de enzimas pancreáticas estuvieron dentro del rango normal, el test de glicemia capilar fue 250 miligramos por decilitro, con osmolaridad sérica conservada y sin acidosis metabólica en la gasometría arterial. Se controló el dolor abdominal y la cefalea con analgesia endovenosa, la presión arterial se normalizó con terapia antihipertensiva y se administró insulina cristalina para la corrección de la glicemia. Se solicitó una tomografía abdominal sin contraste, la cual evidenció una tumoración de densidad mixta de 98 por 82 milímetros aparentemente dependiente de epiplón. El paciente fue referido a un hospital de mayor complejidad para estudio diagnóstico y manejo especializado.
Durante su internamiento no presentó nuevos episodios de crisis hipertensivas, pero persistió el dolor abdominal aunque de menor intensidad respecto al episodio previo. Se agregó además un cuadro de tos productiva y fiebre durante dos días, sin compromiso hemodinámico ni de la oxigenación.
Se realizaron estudios de extensión en búsqueda de enfermedad tumoral a distancia, los que incluyeron tomografía con contraste a nivel cerebral, torácico, abdominal y pélvico. En dichos estudios se evidenció una gran masa heterogénea de 100 milímetros (diámetro longitudinal) por 82 milímetros (diámetro transversal) por 83 milímetros (diámetro anteroposterior), que contactaba superiormente con la cola del páncreas y por debajo con el borde externo del yeyuno proximal; la que comprimía la cara anterior del riñón izquierdo. También se identificó un nódulo heterogéneo en la glándula adrenal derecha de 25 por 23 milímetros (Figura 1).
Figura 1. TomografĒa abdominal con contraste.
A nivel cerebral no se evidenciaron lesiones (Figura 2). No obstante, en la región cervical se halló un nódulo en el lóbulo tiroideo derecho y a nivel del tórax se encontró un infiltrado pulmonar sugestivo de proceso inflamatorio en lóbulo superior derecho (Figura 3). Por este motivo recibió un curso de antibioticoterapia con cefepime a dosis de dos gramos cada ocho horas durante 7 días como tratamiento de neumonía intrahospitalaria, consiguiendo la remisión del cuadro febril mencionado.
Figura 2. TomografĒa cerebral con contraste.
Figura 3. TomografĒa de t¾rax sin contraste.
Posteriormente, el paciente fue hospitalizado en el servicio de medicina interna donde se le realizó una resonancia magnética abdominal, la cual confirmó el origen adrenal izquierdo de la tumoración antes mencionada (Figura 4).
Figura 4. Resonancia magnķtica de abdomen potenciada en T1 y T2 respectivamente.
Ante la sospecha clínica de feocromocitoma bilateral se solicitó el estudio de metanefrinas y ácido vanilmandélico en orina de 24 horas, los cuales fueron positivos. Asimismo, se realizó biopsia por aspiración con aguja fina guiada por ultrasonido de la formación nodular en lóbulo tiroideo derecho, cuyo resultado de citología mostró presencia de células atípicas de patrón fusocelular en grupos con cromatina granular, sugestivo de carcinoma medular de tiroides. Por ello se midió la presencia de otros marcadores como la calcitonina sérica cuyo valor fue de 252 picogramos por mililitro (valor normal hasta 18,2 picogramos por mililitro). Estos hallazgos fueron compatibles con un cuadro de neoplasia endocrina múltiple tipo II.
Entre otros exámenes de ayuda diagnóstica destaca una hemoglobina glicosilada elevada: 12,5%; el hemograma, urea, creatinina, hepatograma y electrolitos se encontraban dentro de rangos normales; la glucosa sérica en ayunas osciló entre 114 a 216 miligramos por decilitro. Los marcadores tumorales fueron negativos a excepción del antígeno carcinoembrionario: 8,95 nanogramo por mililitro (valor normal: 0-4 nanogramo por mililitro); las baciloscopías en esputo fueron negativas (Tabla 1). Se le realizó esofagoduodenoscopía cuyo hallazgo fue gastritis eritematosa leve en cuerpo y antro.
Tabla 1. Pruebas de laboratorio solicitadas.
Una vez completado el estudio diagnóstico se administró un esquema terapéutico durante 14 días como preparación preoperatoria, iniciando α bloqueo con dosis de terazosina de cinco milígramos cada noche por tres días, para luego complementarlo con atenolol 50 milígramos al día.
Antes de la cirugía programada se llevó a cabo una broncofibroscopía, en la que no se encontraron lesiones endobronqueales. Los cultivos de secreciones bronquiales para gérmenes comunes fueron negativos. Las baciloscopía directas también fueron negativas y se solicitó cultivo de dichas baciloscopías.
Finalmente se realizó la adrenalectomía bilateral, obteniéndose un tumor suprarrenal derecho de 20 gramos, de seis por 3,5 por dos centímetros, con superficie de aspecto multinodular amarilla naranja, de consistencia elástica. También se extrajo un tumor suprarrenal izquierdo con peso de 300 gramos, de 10 por ocho por siete centímetros, con superficie externa de color amarillo naranja, de consistencia elástica que a los cortes seriados mostró superficie heterogénea amarronada con áreas hemorrágicas y quísticas (Figura 5A y Figura 5B), cuyos hallazgos histológicos se encontraron relacionados con feocromocitoma derecho encapsulado y feocromocitoma izquierdo de área medular parcialmente encapsulada, más compromiso focal del borde quirúrgico con signos de atipia. La evolución postoperatoria fue favorable.
Figura 5. Pieza operatoria: glßndula suprarrenal izquierda y derecha res-pectivamente.
Catorce días después se hizo la tiroidectomía total, disección central más disección posterolateral de adenopatías a nivel cervical derecho (grupos IIa, IIb, III, IV, V), luego de lo cual evolucionó sin complicaciones postoperatorias. Lo hallazgos histológicos fueron compatibles con cáncer medular de tiroides sincrónico multicéntrico, con invasión de capsula en ambos lóbulos, con compromiso de ganglios linfáticos regionales del grupo II: 1/15, inmunohistoquímica: Factor de transcripción tiroideo 1 (TTF1): positivo focal, tiroglobulina negativo, calcitonina positivo, cromogranina positivo, con estadiaje patológico: pT1a N1 Mx. La glándula paratiroides mostraba hiperplasia, lo que, sumado al hallazgo del carcinoma medular y feocromocitoma bilateral, fue compatible con un cuadro de neoplasia endocrina múltiple de tipo IIA. En la disección central no se encontraron ganglios.
Cuatro semanas después se obtuvieron resultados del cultivo de la baciloscopía en secreción bronquial, los que fueron positivos para Mycobacterium tuberculosis sensible a izoniacida y rifampicina. Por este motivo, se inició inmediatamente tratamiento con drogas antituberculosas de primera línea, las cuales fueron bien toleradas completando esquema de tratamiento por seis meses. El paciente fue dado de alta del programa de control de tuberculosis en condición de curado.
El feocromocitoma es un tumor de presentación poco frecuente, ya que ocurre en menos del 0,3% de pacientes con hipertensión arterial, con una incidencia anual de aproximadamente 0,8 por 100 000 personas al año[4],[5]. Asimismo, puede presentarse de forma esporádica hasta en 60% de casos, siendo la mayoría de ellos encontrados de forma incidental en estudios de autopsias[1]; mientras que en el 40% restante, suelen estar relacionados con una enfermedad hereditaria familiar como el síndrome de Von Hippel-Lindau, la neoplasia endocrina múltiple de tipo II (NEM 2) y la neurofibromatosis tipo 1[2].
La frecuencia de feocromocitoma asociado a neoplasia endocrina múltiple es alrededor de 50%, siendo entre 30% y 50% de ellos asintomáticos, salvo paroxismos de hipertensión arterial en lugar de valores elevados de forma sostenida[3]. Esto concuerda con el caso de nuestro paciente, donde el hallazgo de crisis hipertensiva, cefalea y sudoración profusa dieron cuenta acerca de esta condición.
El hallazgo de dolor abdominal agudo como el que presentó nuestro paciente, no es un síntoma característico del feocromocitoma, tanto en su forma esporádica como hereditaria[6]. La presentación de dolor abdominal como manifestación inicial de neoplasia endocrina múltiple tipo II, sólo ha sido descrito en el estudio de Evans y colaboradores[7]. En dicho estudio se establece como origen de esta dolencia a la hemorragia adrenal que presentó el feocromocitoma del paciente descrito, lo cual generó una gran irritación del peritoneo parietal. Sin embargo, en el caso de nuestro paciente el dolor abdominal no estuvo relacionado a un sangrado del tumor adrenal, como se pudo comprobar en los estudio de imágenes obtenidos. Postulamos que este síntoma podría haber estado relacionado al gran tamaño tumoral del feocromocitoma ubicado en la glándula adrenal izquierda, el cual medía en promedio 10 centímetros de largo, contactaba con la cola del páncreas y con el yeyuno proximal, lo que a su vez pudo haber generado también una estimulación de las fibras dolorosas a nivel peritoneal.
Se han descrito algunas diferencias importantes entre los casos de feocromocitoma esporádico y los asociados a neoplasia endocrina múltiple de tipo II[8]. Los esporádicos casi siempre son unilaterales. Por el contrario, se ha informado que los feocromocitomas asociados a la neoplasia endocrina múltiple de tipo II son bilaterales entre 30 y 100% de los pacientes aproximadamente. El estudio con la mayor cohorte de pacientes con neoplasia endocrina múltiple de tipo IIA, presentado por Modigliani y colaboradores[9], nos muestra que la bilateralidad del feocromocitoma en estos pacientes puede llegar a ser hasta del 67%. Esta condición también se ve reflejada en los hallazgos de nuestro paciente, donde no hubo diferencias histológicas entre los tumores resecados luego de la adrenalectomía bilateral a la que fue sometido.
Los feocromocitomas vinculados a la neoplasia endocrina múltiple de tipo II tienden a manifestarse de forma más temprana en comparación a las formas esporádicas, teniendo como edad media de presentación desde los 25 a los 32 años, dependiendo de la mutación genética que padezcan en el protooncogén rearranged during transfection (RET)[10]. La Sociedad Americana de Endocrinología y Metabolismo recomienda en sus guías de práctica clínica que los pacientes con feocromocitoma sean sometidos a pruebas genéticas para ver no solo la probabilidad de variantes familiares vinculadas a mutaciones de este protooncogen, sino también la asociación con otras enfermedades como el Von Hippel-Lindau[11]. En el caso de esta última, se describen lesiones de tipo hemangioblastomas a nivel del sistema nervioso central, lo cual no fue evidenciado en la tomografía cerebral de nuestro paciente. Una limitación de este reporte de caso es que no se pudo llevar a cabo el estudio genético de dicha condición.
Los feocromocitomas asociados a la neoplasia endocrina múltiple de tipo II suelen presentar una corta duración de síntomas antes del diagnóstico y carecen de un gran tamaño tumoral, teniendo como único denominador común con los casos esporádicos los paroxismos de hipertensión arterial[12]. El caso presentado coincide con dos de estos aspectos ya que se trató de un paciente adulto joven, con sintomatología de poco tiempo de evolución que presentó un episodio de hipertensión arterial asociado a dolor abdominal. La única diferencia estuvo en relación con el tamaño tumoral, ya que el feocromocitoma de la glándula adrenal izquierda alcanzó los 10 centímetros.
La presentación clínica y las manifestaciones del carcinoma medular de tiroides asociado a neoplasia endocrina múltiple de tipo II son similares a aquellos esporádicos, excepto que este último generalmente se presenta más tarde en la vida[13]. Los síntomas más comunes son la presentación de un nódulo tiroideo solitario o linfadenopatía cervical, tal como se reporta en la ecografía tiroidea realizada a nuestro paciente.
Entre los pacientes con neoplasia endocrina múltiple de tipo II, prácticamente todos desarrollan carcinoma medular de tiroides, a menudo con una incidencia máxima en la tercera década de la vida. En cuanto a las características tumorales, se trata de una neoplasia multicéntrica que se concentra en el tercio superior de la glándula tiroides, lo cual refleja la distribución normal de las células parafoliculares productoras de calcitonina. Por otra parte, el carcinoma medular de tiroides suele anteceder al diagnóstico de feocromocitoma en los casos de neoplasia endocrina múltiple de tipo II[14]. Sin embargo, vemos que nuestro paciente debutó con síntomas del tumor adrenal antes mencionado y que el hallazgo de la neoplasia tiroidea se basó en el estudio citológico del nódulo tiroideo encontrado de forma incidental en la tomografía solicitada. Una vez resecada la glándula tiroides, se comprobó mediante histología el carácter multicéntrico antes descrito, además de encontrar una hiperplasia de las glándulas paratiroides; lo cual es compatible con una neoplasia endocrina múltiple de tipo IIA.
La presencia de tuberculosis pulmonar en nuestro paciente responde a múltiples factores. El primero de ellos, y uno de los más importantes, es la procedencia del paciente (Pucallpa, ciudad capital del departamento de Ucayalli, Perú). Según la Dirección General de Epidemiología del Ministerio de Salud de Perú, la tasa de incidencia de tuberculosis en Ucayalli para el año 2015 fue de 131 casos por cada 100 000 habitantes, lo cual supera ampliamente a la incidencia de casos a nivel nacional (87,6 por 100 000 habitantes)[15]. Adicionalmente, si bien se desconoce con exactitud cuál es la relación exacta entre la ocurrencia de tuberculosis y la presentación de una neoplasia endocrina múltiple, se postula que la inmunosupresión causada por el efecto sistémico de una patología da lugar a la adquisición de una nueva infección primaria. Son muy pocos los casos publicados en la literatura científica acerca de la relación entre tuberculosis y feocromocitoma, siendo uno de los más importantes el presentado por Roth y Kvale[16], donde se describe a un paciente con feocromocitoma intratorácico asociado a tuberculosis pulmonar; además del presentado por Toure y colaboradores[17], en el cual se describe a un paciente con feocromocitoma adrenal asociado a tuberculosis sistémica. Un tercer caso es el descrito por Amita K y colaboradores[18], donde el paciente padecía linfadenitis tuberculosa asociado a feocromocitoma lumbar. El escenario clínico que presentamos es inusual, dado que el diagnostico de tuberculosis pulmonar se llevó a cabo en el contexto de una neoplasia endocrina múltiple de tipo IIA, y no como parte de un feocromocitoma esporádico, lo que difiere con los casos reportados en la literatura médica.
Para nuestro conocimiento, reportamos el primer caso de asociación entre neoplasia endocrina múltiple de tipo IIA y tuberculosis pulmonar. La presentación de feocromocitoma en el contexto de esta neoplasia puede estar acompañado no sólo de síntomas clásicos como cefalea, sudoración y crisis hipertensiva, sino también por manifestaciones atípicas como dolor abdominal agudo. Por otra parte, esta patología suele presentarse a edades más tempranas, ser bilateral y preceder a la presentación del carcinoma medular de tiroides.
Agradecimientos
Agradecemos al Instituto de Evaluación de Tecnologías en Salud e Investigación (IETSI) de la Seguridad Social de Perú (EsSalud) por el soporte para el desarrollo de este manuscrito a través de su Programa de Mentoring.
Aspectos éticos
El consentimiento informado solicitado por Medwave, ha sido firmado por el paciente; una copia de este fue remitido a la dirección editorial de la Revista.
Declaración de conflictos de interés
Los autores han completado el formulario de declaración de conflictos de intereses del ICMJE, y declaran no haber recibido financiamiento para la realización del reporte; no tener relaciones financieras con organizaciones que podrían tener intereses en el artículo publicado, en los últimos tres años; y no tener otras relaciones o actividades que podrían influir sobre el artículo publicado. Los formularios pueden ser solicitados contactando al autor responsable o a la dirección editorial de la Revista.
Declaración de financiamiento
Los autores declaran que no hubo fuentes de financiación externas.
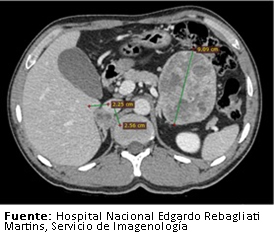 Figura 1. TomografĒa abdominal con contraste.
Figura 1. TomografĒa abdominal con contraste.
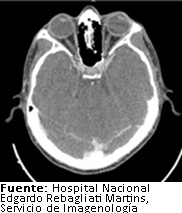 Figura 2. TomografĒa cerebral con contraste.
Figura 2. TomografĒa cerebral con contraste.
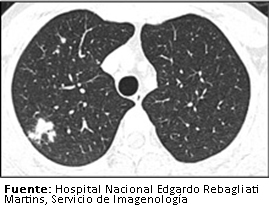 Figura 3. TomografĒa de t¾rax sin contraste.
Figura 3. TomografĒa de t¾rax sin contraste.
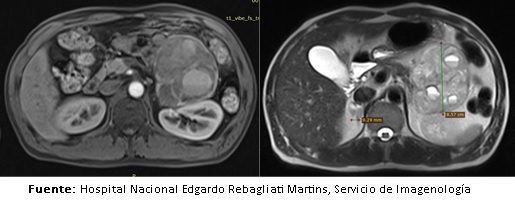 Figura 4. Resonancia magnķtica de abdomen potenciada en T1 y T2 respectivamente.
Figura 4. Resonancia magnķtica de abdomen potenciada en T1 y T2 respectivamente.
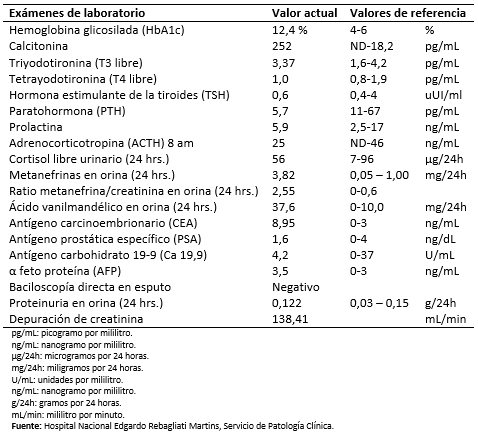 Tabla 1. Pruebas de laboratorio solicitadas.
Tabla 1. Pruebas de laboratorio solicitadas.
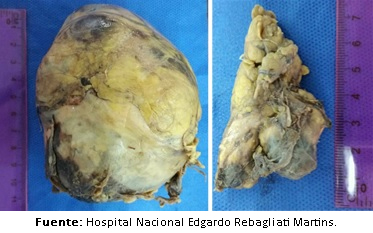 Figura 5. Pieza operatoria: glßndula suprarrenal izquierda y derecha res-pectivamente.
Figura 5. Pieza operatoria: glßndula suprarrenal izquierda y derecha res-pectivamente.
 Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Pheochromocytoma is a catecholamine-producing neoplasm that may occur sporadically or associated with hereditary diseases, such as multiple endocrine neoplasia. The classic symptoms are headache, sweating, and palpitations and are attributed to the sympathetic nervous system activity, usually presenting as paroxysms. On the other hand, pulmonary tuberculosis is an infectious disease considered a public health problem in many countries, whose incidence depends on risk factors such as immunosuppression. It is well known that endocrine-tumor diseases such as multiple endocrine neoplasia can predispose to chronic inflammation and immunosuppression. We report the case of a 38-year-old male patient who had an episode of arterial hypertension and abdominal pain as the first symptoms of a pheochromocytoma associated with multiple endocrine neoplasia type 2A. The patient developed pulmonary tuberculosis simultaneously, but we managed to treat both entities and achieve a favorable clinical course.
Autores:
Josķ Lavalle-MartĒnez[1], Mario Sußrez-Montalvo[2]
Citaci¾n: Lavalle-MartĒnez J, Sußrez-Montalvo M. Pheochromocytoma in multiple endocrine neoplasia 2A associated with pulmonary tuberculosis presenting as abdominal pain: a case report and literature review. Medwave 2018;18(7):e7320 doi: 10.5867/medwave.2018.07.7320
Fecha de envĒo: 18/7/2018
Fecha de aceptaci¾n: 5/10/2018
Fecha de publicaci¾n: 9/11/2018
Origen: no solicitado
Tipo de revisi¾n: con revisi¾n por cuatro pares revisores externos, a doble ciego
Nos complace que usted tenga interķs en comentar uno de nuestros artĒculos. Su comentario serß publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la direcci¾n editorial considera que su comentario es: ofensivo en alg·n sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas polĒticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisi¾n por pares.
A·n no hay comentarios en este artĒculo.
Para comentar debe iniciar sesi¾n
Medwave publica las vistas HTML y descargas PDF por artĒculo, junto con otras mķtricas de redes sociales.
Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc. 1981 Jun;56(6):354-60. | PubMed |Else T. 15 YEARS OF PARAGANGLIOMA: Pheochromocytoma, paraganglioma and genetic syndromes: a historical perspective. Endocr Relat Cancer. 2015 Aug;22(4):T147-59. | CrossRef | PubMed |Rodriguez JM, Balsalobre M, Ponce JL, RĒos A, Torregrosa NM, Tebar J, Parrilla P. Pheochromocytoma in MEN 2A syndrome. Study of 54 patients. World J Surg. 2008 Nov;32(11):2520-6. | CrossRef | PubMed |Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma. A review of the literature and report of one institution's experience. Medicine (Baltimore). 1991 Jan;70(1):46-66. | PubMed |Beard CM, Sheps SG, Kurland LT, Carney JA, Lie JT. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc. 1983 Dec;58(12):802-4. | PubMed |Raue F, Frank-Raue K, Grauer A. Multiple endocrine neoplasia type 2. Clinical features and screening. Endocrinol Metab Clin North Am. 1994 Mar;23(1):137-56. | PubMed |Evans JP, Bambach CP, Andrew S, Dwight T, Richardson AL, Robinson BG, et al. MEN type 2a presenting as an intra-abdominal emergency. Aust N Z J Surg. 1997 Nov;67(11):824-6. | PubMed |Webb TA, Sheps SG, Carney JA. Differences between sporadic pheochromocytoma and pheochromocytoma in multiple endocrime neoplasia, type 2. Am J Surg Pathol. 1980 Apr;4(2):121-6. | PubMed |Modigliani E, Vasen HM, Raue K, Dralle H, Frilling A, Gheri RG, et al. Pheochromocytoma in multiple endocrine neoplasia type 2: European study. The Euromen Study Group. J Intern Med. 1995 Oct;238(4):363-7. | PubMed |Thosani S, Ayala-Ramirez M, Palmer L, Hu MI, Rich T, Gagel RF, et al. The characterization of pheochromocytoma and its impact on overall survival in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. 2013 Nov;98(11):E1813-9. | CrossRef | PubMed |Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014 Jun;99(6):1915-42. | CrossRef | PubMed |Pomares FJ, Ca±as R, Rodriguez JM, Hernandez AM, Parrilla P, Tebar FJ. Differences between sporadic and multiple endocrine neoplasia type 2A phaeochromocytoma. Clin Endocrinol (Oxf). 1998 Feb;48(2):195-200. | PubMed |Wells SA Jr, Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015 Jun;25(6):567-610. | CrossRef | PubMed |Kinlaw WB, Scott SM, Maue RA, Memoli VA, Harris RD, Daniels GH, et al. Multiple endocrine neoplasia 2A due to a unique C609S RET mutation presents with pheochromocytoma and reduced penetrance of medullary thyroid carcinoma. Clin Endocrinol (Oxf). 2005 Dec;63(6):676-82. | PubMed |Alarc¾n V, Alarc¾n E, Figueroa C, Mendoza-Ticona A. [Tuberculosis in Peru: epidemiological situation, progress and challenges for its control]. Rev Peru Med Exp Salud Publica. 2017 Apr-Jun;34(2):299-310. | CrossRef | PubMed |Roth GM, Flock EV, Bollman JL, Kvale WF. Evaluation of the pharmacologic and chemical tests as an aid to diagnosis of pheochromocytoma. Angiology. 1959 Dec;10:426-30. | PubMed |Toure P, Leye A, Leye Y, Diop M, Diop M, Elfajri S, et al. Metastases Of Pheochromocytoma Or Multifocal Tuberculosis? Difficulty Of Diagnosis, A Case Report. The Internet Journal of Endocrinology. 2010;6(2):1-4 | Link |






 Estudios originales
Estudios originales