Revista Biomédica Revisada Por Pares

 Para Descargar PDF debe Abrir sesión.
Para Descargar PDF debe Abrir sesión.
Palabras clave: pulmonary alveolar proteinosis, bronchoalveolar lavage, interstitial lung disease
INTRODUCCIÓN
La proteinosis alveolar pulmonar es una enfermedad intersticial difusa poco frecuente, en la cual se produce obstrucción alveolar, debido al acúmulo de surfactante pulmonar.
REPORTE DEL CASO
Varón de 30 ańos de edad, presentó disnea progresiva y tos seca de un ańo de evolución. Antecedente personal: estibador de cańa de azúcar. Presentó infecciones respiratorias recurrentes. Al examen físico se encontró cianosis, crepitantes difusos bilaterales y subcrepitantes en bases pulmonares. En la tomografía torácica con contraste se encontró un patrón de "empedrado loco". Se realizó videobroncoscopia con lavado broncoalveolar, aspirando material lechoso, espumoso y mucoso. Por citología se encontró linfocitos y material eosinofílico proteináceo acelular. El estudio anatomopatológico de la biopsia transbronquial reveló proteinosis alveolar pulmonar. El paciente reunió los criterios para tratamiento con lavado broncoalveolar total. Luego de este procedimiento, evolucionó favorablemente.
CONCLUSIÓN
La proteinosis alveolar pulmonar constituye una enfermedad importante a considerar, por el desafío diagnóstico y terapéutico que representa.
La proteinosis alveolar pulmonar es una enfermedad intersticial difusa infrecuente, caracterizada por la obstrucción alveolar debida al acúmulo de surfactante pulmonar en el espacio alveolar. Esto genera insuficiencia respiratoria y alteraciones en el intercambio gaseoso pulmonar [1],[2],[3]. Fue descrita por primera vez por Rosen, Castleman y Liebow en 1958 [4]. Su incidencia anual y prevalencia es entre 0,36 y 0,49 hasta 3,7 y 6,2 casos por millón de personas. Presenta un rango de razón masculino/femenino entre 2,1 y 2,7 a uno [5],[6]. Es más frecuente en adultos entre 20 y 50 años, aunque puede afectar a individuos de diferentes edades [7],[8].
Fisiológicamente el surfactante pulmonar reduce la tensión superficial, previene el colapso alveolar y defiende al huésped frente a patógenos microbianos. Es sintetizado y secretado por células epiteliales alveolares tipo II, siendo catabolizado y eliminado en los macrófagos alveolares. El catabolismo del surfactante en macrófagos alveolares requiere la presencia del factor estimulante de la colonia de los granulocitos. Una alteración a este nivel permite el acúmulo alveolar de este material lipoproteináceo, producto de la disminución de su aclaramiento macrofágico, ello explicaría la fisiopatología de la enfermedad [1],[8].
El inicio del cuadro clínico es insidioso, con un curso subagudo indolente que retrasa el diagnóstico [6]. Los síntomas son inespecíficos y son disnea de esfuerzo con o sin tos. Algunos pacientes presentan pérdida de peso, hemoptisis o fiebre. El examen físico es por lo general normal, aunque puede haber crepitantes, acropaquias y cianosis [5],[6],[9]. El diagnóstico se sospecha por el cuadro clínico y los hallazgos radiológicos. Éste se confirma por fibrobroncoscopia con lavado broncoalveolar en los que se encuentre líquido de aspecto lechoso, espumoso, junto con cuerpos amiloides en el examen citológico [1],[6].
Como primera opción, el algoritmo terapéutico en pacientes sintomáticos incluye al lavado broncoalveolar total. Cuando éste es exitoso, se produce una rápida mejoría clínica y radiológica. Sin embargo, son comunes las recaídas y se requiere repetir dicho procedimiento. Otra opción es administrar factor estimulante de colonias de los granulocitos cuando no hay respuesta al primer tratamiento o el paciente no tolera el procedimiento [1],[9],[10]. El pronóstico a largo plazo de estos pacientes es incierto. No obstante, algunos evolucionan de manera estable. Más aún, algunos de ellos presentan mejoría sin tratamiento alguno. Se ha descrito tasas de supervivencia entre 65 y 75% luego de cinco a 10 años del diagnóstico [9]. A continuación se presenta un caso de proteinosis alveolar pulmonar, una enfermedad intersticial difusa, poco frecuente e interesante por el desafío diagnóstico y terapéutico que nos plantea.
Paciente varón, de 30 años de edad, procedente de Chocope. Acudió al Servicio de Emergencia del Hospital Regional Docente de Trujillo, Perú, por presentar disnea progresiva y tos esporádica no productiva de un año de evolución. Tres meses antes del ingreso se agregó sensación de alza térmica. Tres días antes del ingreso presentó empeoramiento de la disnea, por lo cual decidió acudir al servicio de emergencia de dicho hospital.
Dentro de los antecedentes personales se considera que el paciente trabajó como futbolista profesional desde los 19 hasta los 25 años de edad. Hace dos años se desempeñó como estibador de caña de azúcar por un período de seis meses. Asimismo, en lo que respecta a sus antecedentes patológicos, presentó infecciones respiratorias recurrentes tratadas con antibióticos, con resolución parcial de los cuadros infecciosos y pérdida de peso cuantificada de cinco kilogramos en el último año. No hubo antecedente de tuberculosis pulmonar, tabaquismo y alcoholismo. Por otro lado, en lo que concierne a sus antecedentes familiares, su abuelo paterno hace 32 años tenía el diagnóstico de asma bronquial.
Al examen físico, los signos vitales presentaron los siguientes indicadores: presión arterial 120/70 milímetros de mercurio; frecuencia cardiaca 102 latidos por minuto; frecuencia respiratoria 31 respiraciones por minuto; temperatura axilar 38,5 grados Celsius; saturación de oxígeno 88%; fracción inspirada de oxígeno 40%. Se observó un paciente cianótico, que ventilaba espontáneamente en posición semisentada, en regular estado de hidratación y nutrición. A nivel respiratorio, el frémito vocal estaba disminuido en ambos campos pulmonares, había crepitantes difusos bilaterales y subcrepitantes en las bases de ambos campos pulmonares. El resto del examen físico no presentaba alteraciones. En los exámenes de laboratorio, el hemograma y perfil bioquímico se encontraban normales. La enzima lactato deshidrogenasa sérica estaba en 1018 microgramos por decilitro. Los anticuerpos antinucleares y anticuerpos anti DNA de doble cadena eran negativos.
En el análisis de gases arteriales, el cual fue llevado a cabo con porcentaje fraccional de oxígeno inspirado de 21%, se encontró presión parcial de anhídrido carbónico de 26 milímetros de mercurio; presión parcial de oxígeno de 60 milímetros de mercurio; concentración de bicarbonato 17 micromoles por litro; presión parcial de oxígeno/fracción inspirada de oxígeno de 69; y gradiente alveolo-arterial de oxígeno de 531. Asimismo, en el examen directo del esputo con la coloración de Ziehl-Neelsen, no se encontraron bacilos ácido-alcohol resistentes. El test de ELISA para VIH resultó no reactivo. En los estudios de imagen solicitados se observaron infiltrados irregulares, heterogéneos y bilaterales en la radiografía de tórax (Figura 1) y un patrón de “empedrado loco” o crazy paving en la tomografía axial computarizada de tórax, con contraste (Figura 2).
Figura 1. Radiografía de tórax al ingreso.
Figura 2. Tomografía axial computarizada de tórax con contraste durante la hospitalización.
Durante la hospitalización inicial, recibió tratamiento antibiótico con ceftriaxona de dos gramos cada 24 horas por vía endovenosa durante siete días. Posteriormente, se suministró clindamicina de 600 miligramos cada ocho horas por vía endovenosa durante 10 días, y dos tabletas cada ocho horas vía oral por 14 días de cotrimoxazol de 800/160 miligramos. Además, se indicó corticoterapia sistémica con prednisona de 40 miligramos cada 24 horas vía oral por 14 días, presentando mejoría parcial del cuadro clínico.
Se realizó videofibrobroncoscopia diagnóstica, la que evidenció signos inflamatorios agudos en lóbulo superior derecho y lóbulo medio. Además se procedió a un lavado broncoalveolar, aspirando material lechoso, espumoso y mucoso (Figura 3). No hubo anomalías endobronquiales. Por citología se encontraron linfocitos y material eosinofílico proteináceo acelular, que ocupaba algunos espacios alveolares. El estudio anatomopatológico de la biopsia transbronquial informó proteinosis alveolar pulmonar, positivo para la coloración ácido peryódico de Schiff y negativo para la coloración Groccot.
Al tener dicho diagnóstico, el paciente fue nuevamente hospitalizado pues reunía los criterios para ser sometido a lavado broncoalveolar total. Este procedimiento se realizó primero en forma parcial en sala de operaciones y fue continuado en la unidad de cuidados intensivos, con lavado broncoalveolar segmentario en tres oportunidades más. Con posterioridad, recibió ceftazidima de dos gramos cada ocho horas por vía endovenosa durante 10 días; n-acetilcisteina de 600 miligramos cada 12 horas vía oral por 15 días y fisioterapia respiratoria por 10 días. El paciente presentó mejoría del cuadro clínico. Actualmente, permanece estable y acude a sus controles por consultorio externo del servicio de neumología.
Figura 3. Videofibrobroncoscopía.
La proteinosis alveolar pulmonar es una enfermedad rara y se caracteriza por la acumulación de surfactante en los alvéolos. Esto lleva a insuficiencia respiratoria, disfunción de células mieloides y un aumento del riesgo de infecciones respiratorias [11],[12]. La muy baja prevalencia de esta enfermedad fue reportada por Huaringa y Francis [2], quienes realizaron una revisión de la literatura mundial, recopilando 363 casos entre los años 1988 y 2014 en todo el planeta [2]. En Perú existe solamente un caso reportado por Portugal y colaboradores [13], estudiado, diagnosticado y tratado mediante interconsultas realizadas a Pulmonary Consultants Of Palm Beach, Florida, Estados Unidos, en febrero del año 1999 [13].
Existen tres formas principales de esta proteinosis alveolar pulmonar: autoinmune o primaria, secundaria y congénita. En ellas es común la alteración de la eliminación del agente tensioactivo por los macrófagos alveolares, lo que ocasiona su retención en la vía aérea terminal y en el espacio alveolar [5],[9],[10]. Basándonos en la fisiopatología, la proteinosis alveolar pulmonar puede ser causada por la interrupción de la señalización del factor estimulante de colonias de los granulocitos debido a un alto nivel de autoanticuerpos anti factor estimulante de estas colonias de granulocitos (forma autoinmune), la que representa el 90% de los casos. También puede producirse por mutaciones genéticas en los receptores α y β del receptor del factor estimulante de colonias de los granulocitos (forma hereditaria); o por una enfermedad subyacente que afecta a la función y/o número de los macrófagos alveolares (forma secundaria). Esta última puede ocurrir como consecuencia de trastornos clínicos subyacentes como neoplasias malignas hematológicas, inhalación de polvo, tóxicos, humo, gases, inmunosupresión infecciosa o farmacológica [1],[5],[8].
En nuestro paciente probablemente estamos ante una forma secundaria, por la exposición a contaminantes durante su desempeño laboral como estibador de caña. Sin embargo, no es un factor determinante, por falta de estudios que establezcan el tiempo necesario de exposición a un contaminante en específico para adquirir dicha enfermedad. Por otro lado, no podemos excluir que sea una forma autoinmune, una presentación más frecuente de la enfermedad [8].
El curso clínico es variable, pudiendo encontrar pacientes asintomáticos. Hasta casos con insuficiencia respiratoria. Asimismo, los hallazgos clínicos no son específicos, siendo la tos y la disnea los síntomas más frecuentes [7]. Sin embargo, los pacientes pueden presentar fiebre y pérdida de peso, existiendo casos excepcionales de pacientes con hemoptisis. Al examen clínico, los signos más frecuentes constituyen la cianosis, hipocratismo digital y crepitantes bilaterales [8]. En un estudio realizado en China por Mo y colaboradores [5] en el Primer Hospital Afiliado de la Universidad de Medicina de Wenzhou, se revisaron 11 casos de pacientes (siete hombres y cuatro mujeres) con proteinosis alveolar pulmonar. Los síntomas clínicos comunes fueron disnea progresiva de inicio gradual (91%), tos (73%), fiebre (4%) y dolor torácico (18%). El 9% de los pacientes se encontraron asintomáticos. A la exploración física se encontraron crepitantes en 55% de los pacientes, hipocratismo digital en el 45%, cianosis en 18%, y 18% con examen físico normal [5]. El presente caso reportado presentó como síntomas tos, disnea y pérdida de peso; y como signos cianosis, fiebre y crepitantes bilaterales.
Dentro de los estudios imagenológicos usados para el diagnóstico, se encuentra la radiografía de tórax. En ella se aprecian infiltrados nodulares perihiliares bilaterales, con áreas de consolidación, a veces confluentes de predominio en bases pulmonares, mal definidos en mosaico y en vidrio esmerilado simulando “alas de mariposa”, que aparentan una neumonía atípica o un edema pulmonar masivo. En la tomografía axial computarizada de tórax de alta resolución, se observan opacidades difusas en vidrio despulido con sobreposición del engrosamiento septal intra e interlobulillar, patrón conocido como “empedrado loco” o crazy-paving, el cual es característico de esta enfermedad [7],[10],[11]. Ambos hallazgos radiográficos y tomográficos fueron evidenciados dentro de los estudios imagenológicos realizados en el paciente.
Para confirmar el diagnóstico es necesario realizar la biopsia transbronquial por fibrobroncoscopia, donde se observa la arquitectura del parénquima pulmonar conservada. Los hallazgos más notables aquí son el relleno del espacio alveolar con un material acelular granular, amorfo y eosinofílico, que generalmente se tiñe de manera positiva ante el ácido peryódico de Schiff; y la hiperplasia de los neumonocitos de tipo 2 en los septos alveolares. Además, al llevarse a cabo el lavado broncoalveolar, el encontrar líquido opaco de aspecto lechoso y espumoso es patognomónico [1],[9],[14],[15], tal como ocurrió en el presente caso reportado.
El diagnóstico diferencial incluye aquellos trastornos con manifestaciones radiológicas similares. Estos incluyen infección pulmonar por Pneumocystis jirovecii y Mycoplasma sp, edema pulmonar cardiogénico y no cardiogénico, neumonía lipoidea, reacciones de hipersensibilidad relacionadas a fármacos, neumonía organizante, neumonía intersticial aguda y daño alveolar difuso superpuesto a neumonitis intersticial usual, los síndromes de hemorragia alveolar y el antes llamado carcinoma broncoalveolar mucinoso [16].
El tratamiento estándar más aceptado es el lavado broncoalveolar total, con el fin de extraer el material proteináceo que ocupa el espacio aéreo, siendo diagnóstico y terapéutico. Esta técnica está indicada cuando hay un diagnóstico histológico definitivo, presión parcial alveolar de oxígeno menor de 65 milímetros de mercurio, gradiente alvéolo arterial mayor de 40 milímetros de mercurio, presencia de un cortocircuito mayor de 12% y disnea en reposo [7],[11]. Nuestro paciente recibió dicho tratamiento por cumplir con los criterios antes mencionados, mostrando mejoría inmediatamente. Existen alternativas de tratamientos como la administración factor estimulante de colonias de los granulocitos subcutáneo o inhalado, la plasmaféresis y rituximab [8],[10]. Estos se administran cuando hay falta de respuesta o el paciente no tolera el tratamiento convencional. No fue necesario aplicar estos tratamientos en nuestro paciente.
El pronóstico en estos pacientes resulta incierto y los factores que influyen en el mismo son desconocidos. En diferentes estudios se han establecido tres patrones de evolución de la enfermedad: aquellos que permanecen estables, a pesar de presentar episodios recurrentes sintomáticos; los que tienen mejoría espontánea o responden rápidamente días después del tratamiento convencional; y por último, están quienes cursan con deterioro progresivo de la función respiratoria. El 72% de los casos fallece por insuficiencia respiratoria o por un proceso infeccioso no controlado como ocurre en 18% de los casos, generalmente, durante el primer año luego de realizado el diagnóstico [8]. Nuestro paciente tuvo una buena respuesta clínica al lavado broncoalveolar total, permaneciendo estable y controlándose actualmente de manera ambulatoria.
El presente caso de proteinosis alveolar pulmonar resalta la relación de la enfermedad con la exposición laboral, los signos y síntomas inespecíficos y los hallazgos radiográficos y tomográficos característicos. Cuando estamos ante un tipo de enfermedad pulmonar intersticial difusa, debemos tener en cuenta todos estos elementos para sospechar su diagnóstico.
Además, destaca la utilidad de la biopsia transbronquial obtenida mediante broncoscopía diagnóstica para obtener la confirmación histológica, junto con el lavado broncoalveolar total, para servir de herramienta terapéutica en la mayoría de los casos. Por otro lado, se enfatiza que aún hay muchos aspectos de esta enfermedad que aún no están dilucidados y que ameritarían ser investigados.
Aspectos éticos
El consentimiento informado solicitado por Medwave, ha sido firmado por el paciente; una copia de este fue remitido a la dirección editorial de la Revista.
Declaración de conflictos de intereses
Los autores han completado el formulario de declaración de conflictos de intereses del ICMJE, y declaran no haber recibido financiamiento para la realización del reporte; no tener relaciones financieras con organizaciones que podrían tener intereses en el artículo publicado, en los últimos tres años; y no tener otras relaciones o actividades que podrían influir sobre el artículo publicado. Los formularios pueden ser solicitados contactando al autor responsable o a la dirección editorial de la Revista.
Financiamiento
Los autores declaran que no hubo fuentes de financiación externas.
Agradecimientos
Los autores agradecen a los Dres. José Cárdenas-García, Julio Hilario-Vargas y Tony Chávez-Uceda, por haber revisado cuidadosamente el manuscrito y por sus sugerencias más útiles.
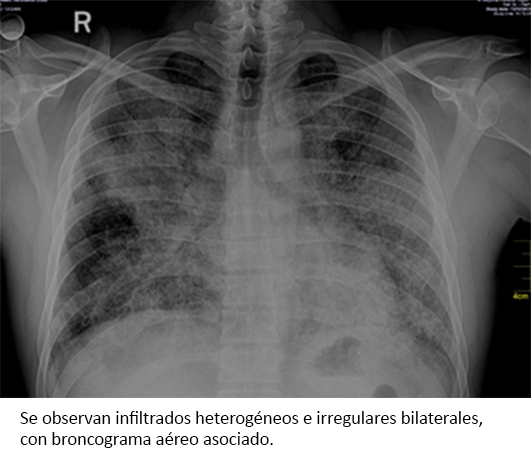 Figura 1. Radiografía de tórax al ingreso.
Figura 1. Radiografía de tórax al ingreso.
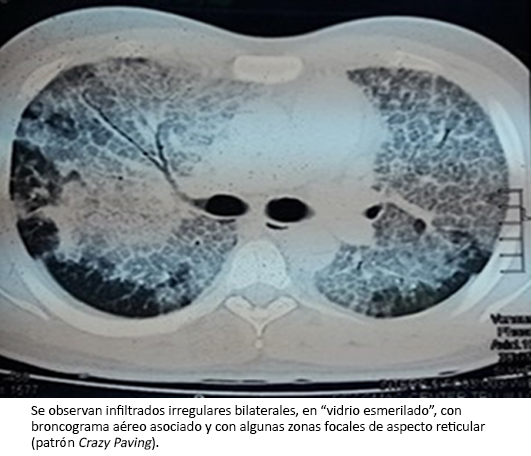 Figura 2. Tomografía axial computarizada de tórax con contraste durante la hospitalización.
Figura 2. Tomografía axial computarizada de tórax con contraste durante la hospitalización.
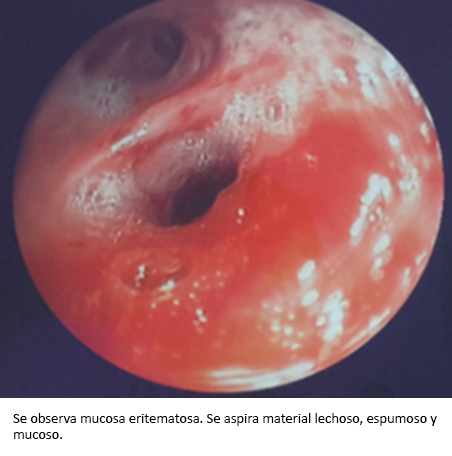 Figura 3. Videofibrobroncoscopía.
Figura 3. Videofibrobroncoscopía.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
INTRODUCTION
Pulmonary alveolar proteinosis is a rare, diffuse interstitial lung disease, characterized by alveolar obstruction due to the accumulation of pulmonary surfactant.
CLINICAL PRESENTATION
A 30-year-old male with progressively worsening dyspnea and non-productive cough for one year. He was a sugar cane plantation worker and had prior recurrent respiratory infections. Physical exam revealed cyanosis, and bilateral coarse and fine rales. Chest computed tomography showed diffuse crazy paving pattern. Bronchoscopy with bronchoalveolar lavage yielded a foamy, thick whitish material. Cytology revealed lymphocytes and acellular proteinaceous eosinophilic material. Transbronchial biopsy confirmed the diagnosis of pulmonary alveolar proteinosis. Patient met criteria for whole lung lavage, responding favorably to this therapy.
CONCLUSION
Pulmonary alveolar proteinosis is a rare lung disease and important to consider due to the diagnostic and therapeutic challenge it represents.
Autores:
Luis Alberto Concepción-Urteaga[1,2], Luis Alejandro Rodríguez-Hidalgo[1,2], Jorge Luis Cornejo-Portella[1,2], Oscar Neri Alquizar-Horna[1,2], Daniel Anderson Aguilar-Villanueva[2], Marcio José Concepción-Zavaleta[2], Mario Gustavo Azańero-Luján[3]
Citación: Concepción-Urteaga LA, Rodríguez-Hidalgo LA, Cornejo-Portella JL, Alquizar-Horna ON, Aguilar-Villanueva DA, Concepción-Zavaleta MJ, et al. Pulmonary alveolar proteinosis: a case report. Medwave 2017 Sep;17(8):e7040 doi: 10.5867/medwave.2017.08.7040
Fecha de envío: 23/6/2017
Fecha de aceptación: 23/8/2017
Fecha de publicación: 14/9/2017
Origen: no solicitado
Tipo de revisión: con revisión por cuatro pares revisores externos, a doble ciego
Nos complace que usted tenga interés en comentar uno de nuestros artículos. Su comentario será publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la dirección editorial considera que su comentario es: ofensivo en algún sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas políticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisión por pares.
Aún no hay comentarios en este artículo.
Para comentar debe iniciar sesión
Medwave publica las vistas HTML y descargas PDF por artículo, junto con otras métricas de redes sociales.
Suzuki T, Trapnell BC. Pulmonary Alveolar Proteinosis Syndrome. Clin Chest Med. 2016 Sep;37(3):431-40. | CrossRef | PubMed |Huaringa AJ, Francis WH. Pulmonary alveolar proteinosis: a case report and world literature review. Respirol Case Rep. 2016 Nov 13;4(6):e00201. | CrossRef | PubMed |McDonnell MJ, Reynolds C, Tormey V, Gilmartin JJ, Rutherford RM. Pulmonary alveolar proteinosis: report of two cases in the West of Ireland with review of current literature. Ir J Med Sci. 2014 Mar;183(1):123-7. | CrossRef | PubMed |Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958 Jun 5;258(23):1123-42. | PubMed |Mo Q, Wang B, Dong N, Bao L, Su X, Li Y, et al. The Clinical Clues of Pulmonary Alveolar Proteinosis: A Report of 11 Cases and Literature Review. Can Respir J. 2016;2016:4021928. | CrossRef | PubMed |Khan A, Agarwal R. Pulmonary alveolar proteinosis. Respir Care. 2011 Jul;56(7):1016-28. | CrossRef | PubMed |Sánchez-Valadez TI, Carrillo-Muńoz A, Valero-Gómez A, Martínez-Pérez S, Pérez-Rosales A, Núńez Pérez-Redondo C, et al. Proteinosis alveolar: Informe de dos casos y comentario bibliográfico. Neumología y cirugía de tórax. 2015;74(4):271-5. | Link |Algarin H, Mendoza DA, Molano D, Claro J. Proteinosis alveolar pulmonar: diagnóstico y manejo en unidad de cuidado intensivo. Reporte de caso. Acta Colombiana de Cuidado Intensivo. 2017;17(1):76-80. | Link |Ernst G, Salvado A, Grynblat P, Tabaj G, Garcia O, Cambursano VH, et al. [Pulmonary alveolar proteinosis: role of GM-CSF Antibodies]. Rev Fac Cien Med Univ Nac Cordoba. 2017;74(1):46-50. | PubMed |Rodríguez J. Tratamiento de la proteinosis alveolar primaria del adulto. Arch Bronconeumol. 2015;51(7):344-9. | Link |Fijołek J, Wiatr E, Opoka L, Rudziński P, Nierebińska M, Szołkowska M, et al. Atypical image of pulmonary alveolar proteinosis - a case report. Pneumonol Alergol Pol. 2015;83(6):453-6. | CrossRef | PubMed |Ul Rehman S, Hammersley JR. Pulmonary Alveolar Proteinosis: Crazy Paving to Whole Lung Lavage. Mayo Clin Proc. 2016 Nov;91(11):1673-674. | CrossRef | PubMed |Portugal J, Torres E, Díaz F, Minauro C. Proteinosis Alveolar Pulmonar. Boletín de la Sociedad Peruana de Medicina Interna. 2000;13(1):47-51. | Link |Munir Z, Khosa MZ, Qazi MY. Pulmonary Alveolar Proteinosis: A Rare Cause of Respiratory Failure. J Coll Physicians Surg Pak. 2015 Jul;25(7):541-2. | CrossRef | PubMed |Yildirim BB, Akgedik R, Akgedik S, Nazaroglu H. Pulmonary alveolar proteinosis in a marble worker. Int J Occup Med Environ Health. 2016;29(5):871-6. | CrossRef | PubMed |Chan ED, King TE. Causes, clinical manifestations, and diagnosis of pulmonary alveolar proteinosis in adults. Walthman, MA: UpToDate; 2017. | Link |






 Estudios originales
Estudios originales