Revista Biomķdica Revisada Por Pares

 Para Descargar PDF debe Abrir sesi¾n.
Para Descargar PDF debe Abrir sesi¾n.
Palabras clave: hereditary angioedema type I, complement C1 inhibitor protein, diagnoses
El angioedema hereditario es una enfermedad rara, de gran heterogeneidad en los sĒntomas, manifestßndose con edema a nivel cutßneo, mucosa gastrointestinal y de laringe/faringe. Aunque existen tres variedades, el tipo I es el mßs frecuente y es provocado por una deficiencia en la sĒntesis del complemento C1 inhibidor. La gravedad de la clĒnica, junto a la baja prevalencia de la enfermedad y la necesidad de un tratamiento especĒfico, hacen que el diagn¾stico y tratamiento de dicha patologĒa sea a·n una asignatura pendiente para el mķdico de familia en atenci¾n primaria. Presentamos el caso de un adolescente var¾n con dķficit de α-1 antitripsina desde los seis meses de edad, con aparici¾n de angioedemas en piernas y brazos a los 11 a±os, diagnosticado de angioedema hereditario tipo I un a±o despuķs. El diagn¾stico definitivo de la enfermedad permiti¾ instaurar un tratamiento adecuado a su patologĒa, que consiste en la prevenci¾n de brotes que puedan comprometer la vida del paciente y, en el caso de que aparezcan, en la administraci¾n del complemento C1 inhibidor.
El angioedema hereditario o edema de Quincke (nombre debido a su descubridor, 1882), es una enfermedad genética caracterizada por edemas circunscritos y deformantes que implican la piel, el tejido subcutáneo, las membranas mucosas y algunas veces las vísceras.
Se trata de una enfermedad rara, de baja prevalencia (entre uno y nueve casos por 100 000 personas) [1]. Se presenta con una clínica de gran heterogeneidad y frecuentemente se diagnostica de forma errónea como enfermedad autoinmune o anafilaxia. La aparición de edema de glotis en 25 a 30% de los casos [2] y un 13% de muertes por asfixia [3], justifican la necesidad de un diagnóstico lo más precoz posible y la implementación de un tratamiento profiláctico que disminuya la aparición de síntomas graves que puedan comprometer la vida del paciente, y en caso de que estos se presenten, la administración del único tratamiento válido para prevenir la mortalidad por esta causa: el inhibidor C1 esterasa. Nos encontramos ante una enfermedad de gran impacto en la calidad de vida del paciente que lo sufre y de sus familiares, tanto por la recurrencia de los síntomas como por la gravedad de los mismos.
Tras una revisión de la literatura existente, hemos encontrado asociaciones descritas entre el angioedema hereditario y enfermedades autoinmunes como la tiroiditis, lupus eritematoso, síndrome de Sjögren o enfermedad inflamatoria intestinal [4], pero ningún otro caso que asocie el déficit de α-1 antitripsina con el déficit de inhibidor C1 esterasa.
Se presenta el caso de un adolescente, natural de Cádiz (España), a cuyos padres se les solicitó el consentimiento informado para su presentación.
Entre sus antecedentes destacamos un déficit de α-1 antitripsina que se manifestó a los nueve meses de edad, tras varios cuadros de bronquiolitis de repetición y en seguimiento por el Hospital Universitario de Puerta del Mar (Cádiz). Los valores de α-1 antitripsina han ido aumentando paulatinamente, sin necesidad de tratamiento específico, con valores que van desde los 76,9 mg/dl en el primer año de vida hasta casi su normalización con 88,6 mg/dl a los 14 años de edad, sin que hasta el momento dicho déficit haya tenido repercusión hepática o gastrointestinal. Actualmente, el joven presenta asma bronquial no alérgico con exacerbaciones periódicas que le provoca disnea a moderados esfuerzos en tratamiento con plusvent® 25/250 mcg aerosol y montelukast 5 mg. Su peso actual es de 52 kilos y su altura 156 centímetros. Presenta un índice de masa corporal de 21,37, encontrándose en el percentil 75.
Entre los antecedentes familiares encontramos que el padre también presenta déficit de α-1 antitripsina sin tratamiento y un primo de la abuela materna falleció por lupus eritematoso sistémico. No existen antecedentes evidentes en la línea materna o paterna de angioedema recurrente. Tanto el paciente como los progenitores están pendientes de estudio genético.
A los 11 años comienza a presentar episodios de angioedemas recurrentes en miembros periféricos, de color rosáceo o blanquecino, no asociados a urticaria, dolorosos a la palpación y que provocan molestias cuando aparecen en las articulaciones. En varias ocasiones ha presentado edemas palpebrales y en región facial, repitiéndose estos episodios cada 30 o 45 días las primeras veces y sin que se asociasen a ninguna causa aparente. Casi todos estos cuadros fueron tratados con esteroides y antihistamínicos, tanto en atención primaria como a nivel hospitalario, encontrando mejoría clínica a partir de las 36 horas con la administración del tratamiento y sin él.
Tras diez meses de evolución presenta un episodio con edema gloso-uvular y faríngeo, tratado de igual forma que en ocasiones anteriores. En estos momentos los brotes han ido aumentado su frecuencia hasta llegar a presentar de dos a tres brotes en un mismo mes.
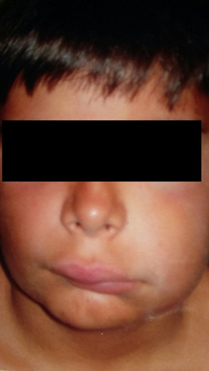
Figura 1. Angioedema facial.
Tras la persistencia, mayor frecuencia (se han descrito diez episodios en seis meses) y gravedad de los episodios, se decide realizar estudio por dermatólogo y alergólogo de zona que descarta enfermedad alérgica o reacción anafiláctica. Esto es demostrado analíticamente con la determinación de inmuloglobulina E (Ig E) sérica dentro de la normalidad.
Entre los hallazgos analíticos destacamos:
Como consecuencia de los hallazgos anteriores, se decide ampliar el estudio analítico con las siguientes determinaciones:
|
Valores encontrados |
Valores de referencia |
|
|
Complemento C3 |
148,20 mg/dl |
90-180 mg/dl |
|
Complemento C4 |
0,90 mg/dl |
10-40 mg/dl |
|
Complemento C1q |
22,30 mg/dl |
10-25 mg/dl |
|
Inhibidor C1 esterasa |
6,14 mg/dl |
22-34 mg/dl |
|
Complemento C5 |
19,90 mg/dl |
4-15 mg/dl |
Los datos clínicos junto con los del laboratorio (niveles bajos de C4, C1q dentro de los valores normales e inhibidor C1 esterasa al 24% de su valor normal), confirmó el diagnóstico de angioedema hereditario tipo I, lo que permitió la introducción de un tratamiento individualizado y adecuado a su patología. Desde ese momento se descartó el uso de corticoides, antihistamínicos y epinefrina para tratar los brotes. Actualmente, el seguimiento del paciente se lleva a cabo por la unidad de alergología del Hospital Universitario Virgen del Rocío (Sevilla).
Desde el inicio de los síntomas hasta el diagnóstico de la enfermedad y la implementación del tratamiento, pasaron 14 meses. Los padres cuantificaron un total de aproximadamente 15 episodios de angioedemas en este periodo. La edad del paciente (12 años) al momento del diagnóstico del angioedema hereditario, hizo que se descartasen los andrógenos atenuados como tratamiento preventivo de los brotes y que se eligiese un antifibrinolítico para la profilaxis a largo plazo. Este consistió en el uso de ácido tranexámico de 500 mg cada 8 horas, lo que le ha permitido disminuir la frecuencia de los episodios y su intensidad.
Desde que se instauró el tratamiento hace 20 meses, la frecuencia de brotes ha disminuido considerablemente, dándose sólo tres incidencias que consistieron en dos episodios de edemas de menor tamaño en brazos y un episodio de edema palpebral sin que ninguno necesitase tratamiento médico y sin que se hayan aparecido edemas en las vías respiratorias altas ni síntomas digestivos. Uno de estos episodios se asoció en el tiempo a un cuadro febril de 48 horas de evolución, mientras que los otros dos no tuvieron causa aparente. Ninguno duró más de 24 horas. Además, se le aportó al paciente Cinryze® (inhibidor de la C1 esterasa humana) para su administración intravenosa en caso de brote agudo importante o como profilaxis a corto plazo en caso de intervención quirúrgica, sin que hasta el momento haya sido necesario su uso.
El tratamiento preventivo con antifibrinolíticos, aunque está siendo bien tolerado por el paciente, obliga a realizarle controles con estudios de la función renal y hepática, junto con revisiones oftalmológicas periódicas para control de retinopatías o glaucoma, sin que hayan aparecido complicaciones hasta este momento. En la actualidad sigue manteniendo la misma dosis de ácido tranexámico desde que se instauró el tratamiento.
El control de los episodios agudos desde el inicio del tratamiento, hizo que tanto los padres como el paciente percibiesen la enfermedad con menos negatividad y con mayores expectativas de salud.
El angioedema recibe por primera vez el nombre de edema angioneurótico en 1882, a manos de Heinrich Quincke. En 1888, William Osler habló de una base hereditaria y en 1917 se identificó su herencia autosómica dominante. Fue en 1963 cuando Donaldson y Evans describieron la causa genética de la enfermedad como una deficiencia del inhibidor sérico del complemento C1 (inhibidor C1 esterasa) [5].
La deficiencia genética del angioedema puede tener un origen hereditario o adquirido y se manifiesta por la ausencia o síntesis insuficiente del inhibidor C1 esterasa [6], necesario para regular la activación de la vía clásica del complemento. El inhibidor C1 esterasa pertenece al sistema inhibidor de las serin-proteasas (serpinas) del plasma humano, igual que otras proteínas como la antitrombina III, la α-2-antiplasmina, la α-1 antitripsina (cuyo déficit también encontramos en nuestro paciente), entre otras.
De forma clásica, se han diferenciado dos variedades del angioedema hereditario. El angioedema hereditario tipo I es la forma más común, representando el 85% de los casos aproximadamente. Es causado por una síntesis deficiente del complemento C1 inhibidor, como ocurre en el caso de nuestro adolescente. El tipo II supone aproximadamente el 15% restante y, aunque el paciente puede presentar unos niveles normales, o incluso elevados, de inhibidor C1 esterasa, funcionalmente son deficientes. A pesar de esta clasificación, se ha descrito una tercera variante, el angioedema hereditario tipo III [7],[8], con una clínica idéntica a los dos anteriores, en el que la concentración y funcionalidad del inhibidor C1 esterasa son normales, y que sólo afecta a mujeres, siendo estrógeno dependiente y relacionándose con el embarazo o uso de anticonceptivos. De este último tipo sólo se han descritos unos pocos casos [9].
Se trata de una enfermedad rara, con una prevalencia baja, entre uno y nueve por cada 100 000 personas, sin encontrar diferencias de género ni predominio de razas [1], exceptuando el angioedema hereditario tipo III que se da en mujeres y está ligado al cromosoma X [5].
A pesar de la tipología de la enfermedad, las manifestaciones clínicas son prácticamente idénticas en cualquiera de sus variantes. Éstas se caracterizan por episodios recurrentes y autolimitados en el tiempo de edemas que afectan al tejido subcutáneo, vísceras y mucosa del tracto gastrointestinal, junto con vías respiratorias altas [10], con gran variabilidad en los síntomas. La gravedad de éstos dependerá del grado de afectación y localización del edema. Generalmente el angioedema cutáneo suele no ser doloroso, no asociado a urticaria, blanquecino y no eritematoso, como el descrito en este adolescente. Otros síntomas que pueden aparecer son edema de glotis y/o faríngeo, disnea, disfagia, distensión abdominal, vómitos, diarrea y estreñimiento, aunque hasta el momento nuestro paciente no ha sufrido síntomas digestivos. También se conoce que ciertas circunstancias como el estrés, procesos infecciosos, la cirugía, intubación y tratamientos invasivos dentales [11], son desencadenantes del brote agudo, por lo que se podría condicionar el episodio febril que padeció el joven a la exacerbación del cuadro.
El angioedema hereditario se debe sospechar ante episodios recurrentes de angioedema subcutáneo sin urticaria, cuadros repetidos de dolor abdominal autolimitados o edema laríngeo de origen no anafiláctico. El diagnóstico de la enfermedad se confirma en función de los niveles séricos de complemento [8],[12] tal y como ocurrió en nuestro caso, ya que el escrutinio es sencillo:
El inicio de las manifestaciones clínicas puede darse a cualquier edad, aunque es más frecuente en la infancia y adolescencia [4] como es el caso de este paciente con presencia de síntomas por primera vez a los once años de edad. En él se evidenciaron niveles séricos de inhibidor C1 esterasa 3,6 veces por debajo del nivel normal con C1q normal y niveles de C4 nueve veces más bajo que el nivel mínimo normal. Estos datos confirman el diagnóstico de angioedema hereditario tipo I.
La necesidad de un tratamiento precoz radica en su importancia para evitar las complicaciones. Tanto la profilaxis como el tratamiento agudo tienen como finalidad disminuir la intensidad del edema, evitar la mortalidad y evitar su impacto emocional negativo [13], objetivos que de momento se han conseguido en el caso del adolescente que reportamos, pasando de 15 episodios en un año a tres episodios una vez instaurado el tratamiento. Este tratamiento incluye:
Nos encontramos ante una enfermedad en la que, aunque el 75% de los pacientes presentan su primer episodio antes de los 15 años, el diagnóstico suele tardar entre ocho [14] y quince años [3] en realizarse. Este retraso cobra especial relevancia ya que en la actualidad, la mortalidad por angioedema hereditario disminuye drásticamente una vez realizado el diagnóstico e iniciado el tratamiento adecuado.
Es importante realizar un buen diagnóstico diferencial, ya que los síntomas digestivos pueden llegar a confundir al profesional con un cuadro de abdomen agudo. En el caso del edema faringolaríngeo puede tratarse como una reacción anafiláctica. Esta situación es bastante grave, ya que en el caso del angioedema hereditario no existe respuesta a corticoides, antihistamínicos ni adrenalina, lo que puede provocar muerte por asfixia con una mortalidad descrita entre el 13 y 30% [3],[4].
Además, el paciente presenta como patología de base déficit de α-1 antitripsina y en la literatura revisada no se han encontrado casos descritos con comorbilidad asociada a angioedema hereditario. Aparentemente, este sería el primer caso reportado, no obstante sí se han descrito en los últimos años asociaciones entre el angioedema hereditario y enfermedades autoinmunes como es el caso del lupus eritematoso sistémico, enfermedad que encontramos en un miembro de la familia.
El angioedema hereditario, aunque es una patología de baja prevalencia, puede llegar a ser mortal si no la diagnosticamos a tiempo, debido a que la clínica nos puede hacer sospechar otras patologías tratándose al paciente de forma errónea. Nuestro paciente sufrió aproximadamente 15 episodios que fueron diagnosticados como urticaria o reacción alérgica en las urgencias de atención primaria y hospitalaria, hasta su correcto diagnóstico. Todo esto nos hace ver la importancia de repasar esta dolencia poco conocida entre los profesionales sanitarios y que puede darse en atención primaria, puerta de todas las patologías, y de fácil diagnóstico si ante la sospecha en presencia de angioedema se realizase una determinación sérica de los complementos.
Aspectos éticos
El consentimiento informado solicitado por Medwave, ha sido firmado por la madre del paciente; una copia de este fue remitido a la dirección editorial de la revista.
Declaración de conflictos de intereses
Los autores han completado el formulario de declaración de conflictos intereses del ICMJE traducido al castellano por Medwave, y declaran no haber recibido financiamiento para la realización del reporte; no tener relaciones financieras con organizaciones que podrían tener intereses en el artículo publicado, en los últimos tres años; y no tener otras relaciones o actividades que podrían influir sobre el artículo publicado. Los formularios pueden ser solicitados contactando al autor responsable o a la dirección editorial de la Revista.
Financiamiento
Los autores declaran que no hubo fuentes de financiación externas.
 Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Hereditary angioedema is a rare disease with great heterogeneity of symptoms such as edema of the skin, gastro-intestinal mucosa and larynx or pharynx. Even though there are three types, the most frequent is type I, which is a result from a deficiency of the complement C1 inhibitor. The severity of its symptoms along with the low prevalence of the disease and the need for appropriate specific treatment make the diagnosis and treatment of the pathology an outstanding subject for the family physician. The present is the case of a male teenager with alpha-1 antitrypsin deficiency since he was six months old, angioedema on arms and legs since 11 years old and diagnosed with hereditary angioedema type I one year after. The definitive diagnosis of the disease enabled an appropriate treatment which consists in preventing outbreaks that may compromise the patient's life and, if they occur, administration of complement C1 inhibitor.
Autores:
Francisca Mu±oz Peralta[1], Eva Buller Viqueira[2], Juana Cabello Pulido[3]
Citaci¾n: Mu±oz Peralta F, Buller Viqueira E, Cabello Pulido J. Hereditary angioedema type I: a case report. Medwave 2016 Ene;16(1):e6378 doi: 10.5867/medwave.2016.01.6378
Fecha de envĒo: 4/11/2015
Fecha de aceptaci¾n: 18/12/2016
Fecha de publicaci¾n: 28/1/2016
Origen: no solicitado
Tipo de revisi¾n: con revisi¾n por un par revisor externo, a doble ciego
Nos complace que usted tenga interķs en comentar uno de nuestros artĒculos. Su comentario serß publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la direcci¾n editorial considera que su comentario es: ofensivo en alg·n sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas polĒticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisi¾n por pares.
Nombre/name: Eva Buller
Fecha/date: 2016-05-09 13:40:39
Comentario/comment:
Me dirijo a la editorial, soy la segunda autora de esta publicaci├│n. Al descargar en PDF el art├Łculo mi segundo apellido esta mal escrito, en cambio aqu├Ł no. El segundo apellido es VIQUEIRA. ┬┐Lo podr├Łan corregir? Un saludo.
Para comentar debe iniciar sesi¾n
Medwave publica las vistas HTML y descargas PDF por artĒculo, junto con otras mķtricas de redes sociales.
Jimenez Saab N, G¾mez Vera J, G¾mez Tiro J, Nieto MartĒnez S, Pliego Reyes C. Angioedema hereditario. Comunicaci¾n de un caso y revisi¾n de la bibliografĒa. Rev Aler Mķx. 2006; 53(1):34-41. | Link |Fernßndez Romero DS, Dimarco P, Malbran A. Angioedema hereditario. Historia familiar y manifestaciones clĒnicas en 58 pacientes. Medicina (Buenos Aires) 2009;69:601-606. | Link |Vives Toledo R, SorlĒ Guerola JV, Sierra Santos L, GarcĒa Ribes M. Angioedema hereditario. Rev ClĒn Med Fam. 2015; Vol 8(1):62-65. | Link |Navarro Ruiz A, Crespo C, Poveda Andrķs JL, Cebollero de Torre A. Algoritmo de diagn¾stico y tratamiento del angioedema hereditario como herramienta para su manejo. Farm Hosp. 2013;37(6):521-529. | CrossRef |Donalson VH, Evans RR. A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C1-esterase. Am J Med. 1963 Jul;35:37-44. | PubMed |Bork K, Barnstedt SE, Kock P, Traupe H. Hereditary Angioedema with normal inhibidor C1 esterasaibitor activity in women. Lancet. 2000 Jul 15;356(9225):213-7. | PubMed |┴lvarez Caro F, DĒaz MartĒn JJ, ┴lvarez Berciano F. Edema angioneur¾tico hereditario en pediatrĒa: consideraciones a prop¾sito de tres casos. Act Ped Esp. 2007;65(6):300-303. | Link |Calvo G¾mez-Rodulfo A, GarcĒa L¾pez JE, Herrero-MorĒn JD, RodrĒguez GarcĒa G, Gonzßlez Guerra F. Angioedema hereditario en pediatrĒa. Bol Pediatr. 2009; 49:16-23. | Link |Past¾ Cardona L, Bordas Orpinell J, Mercadal Orfila G, Pķrez de la Vera A, J¾dar Massanķs R. Profilaxis y tratamiento del angioedema hereditario y adquirido en el HUB; utilizaci¾n del inhibidor de la C1-esterasa. Farm Hosp. 2003;27(6):346-352. | Link |Malbran A, Fernßndez Romero DS, Menķndez A. Angioedema hereditario. GuĒa de tratamiento. Medicina (Buenos Aires) 2012;72:119-123. | Link |Caballero T1, Baeza ML, Caba±as R, Campos A, Cimbollek S, G¾mez-Traseira C, et al. Consensus Statement on the Diagnosis, Management, and Treatment of Angioedema Mediated by Bradykinin. Part II. Treatment, Follow-up, and Special Situations. J Investig Allergol Clin Immunol. 2011;21(6):422-41; quiz 442-3. | PubMed |Rodriguez Labrada M, Surday Berm·dez L, Corrales ┴lvarez M. Enfermedad de Quincke, desafĒo del especialista en otorrinolaringologĒa. Acta Mķdica del Centro. 2014; Vol 8(3). | Link |Huang SW. Result of an online survey of patients with hereditary Angioedema. Allergy Asthma Proc. Allergy Asthma Proc. 2004 Mar-Apr;25(2):127-31. | PubMed |






 Estudios originales
Estudios originales