Revista Biomķdica Revisada Por Pares

 Para Descargar PDF debe Abrir sesi¾n.
Para Descargar PDF debe Abrir sesi¾n.
Este texto corresponde a un trabajo de revisi¾n preparado por sus autores en el desarrollo del Curso y Seminarios de OncologĒa Bßsica, realizado por el Centro de OncologĒa Preventiva y la Escuela de Postgrado de la Facultad de Medicina de la Universidad de Chile entre abril y agosto de 2008. El Director del Curso es el Dr. Josķ Manuel Ojeda.
La leucemia aguda es el cßncer mßs frecuente en la edad pedißtrica: representa 30% de todos los cßnceres infantiles en los Estados Unidos y 40% en Chile, y es la primera causa de mortalidad por cßncer. Esta patologĒa puede tener dos fenotipos: linfoblßstico (LLA) o mieloblßstico (LMA); el primero es el mßs frecuente, ya que corresponde a alrededor de 80% de todos los casos (1-5).
La translocaci¾n cromos¾mica es uno de los mecanismos esenciales en el desarrollo de un tumor y es muy com·n en ambos tipos de leucemia. Esta alteraci¾n determina la activaci¾n de oncogenes mediante la fusi¾n de genes, lo que habitualmente origina productos reguladores de la transcripci¾n. Dentro de los subtipos de leucemia aguda se han descrito asociaciones especĒficas con alteraciones cromos¾micas que se utilizan como factor pron¾stico y para estratificar grupos de riesgo, aspecto importante para definir alternativas de tratamiento (3-5).
Uno de los puntos de quiebre cromos¾mico mßs frecuentes en las leucemias humanas se ubica en el cromosoma 11 banda q23, donde reside el gen de Linaje Leucķmico Mixto (Mixed Lineage Leukemia o MLL, tambiķn llamado ALL-1 o HRX), el cual constituye una de las excepciones dentro de las asociaciones especĒficas; se ha descrito su presencia en mßs de 60 genes fusionados, por lo que se dice que es un oncogen ōpromiscuoö. Estas translocaciones se pueden observar en una amplia gama de patologĒas malignas hematol¾gicas, principalmente en LLA y LMA, asĒ como tambiķn en sĒndromes mielodisplßsicos y en el linfoma de Burkitt (4, 5).
Los rearreglos del gen MLL, que determinan distinto pron¾stico terapķutico seg·n el fenotipo de leucemia, se detectan en 6% de los casos de LLA y 80% de ķstos ocurren en menores de un a±o de edad (leucemia del lactante). Su presencia constituye un factor de mal pron¾stico, que empeora en relaci¾n inversa con la edad del paciente. En la LMA son mßs frecuentes: se ven en14 % de los casos totales y en 65% de los casos en lactantes y confieren un pron¾stico de riesgo intermedio (4, 5).
Tambiķn se ha visto involucrado al gen de MLL en leucemias secundarias a tratamiento, principalmente del tipo LMA en pacientes tratados con inhibidores de la topoisomerasa II como tratamiento de un cßncer primario. Se ha postulado la presencia de mecanismos similares para la leucemia del lactante cuyas madres tuvieron exposici¾n a topoisomerasas II nativas (4).
A continuaci¾n se revisarß distintos aspectos de la relaci¾n entre el gen MLL y la leucemia aguda en pacientes pedißtricos.
El gen MLL se ubica en el cromosoma 11q23, justo despuķs del dominio represor; tiene una extensi¾n de 90 kb y se compone de 38 exones; produce un mRNA de 12 kb que codifica una proteĒna de 3969 aminoßcidos, de 430 kDa de peso molecular y compleja estructura. Esta proteĒna se expresarĒa ampliamente en el embri¾n en desarrollo, donde funcionarĒa como regulador de la transcripci¾n nuclear; en tejidos adultos sus niveles son mĒnimos (5-7).
La proteĒna MLL normalmente es clivada en el citoplasma por la taspasa 1 en los aminoßcidos 2666 (sitio de clivaje 1 o CS1) y 2718 (sitio de clivaje 2 o CS2), generando dos subunidades: MLL-N (300 kDa) y MLL-C (180 kDa). Su funci¾n es acetilar, desacetilar y metilar las histonas de los nucleosomas (8). La proteĒna madura contiene una regi¾n cluster de puntos de quiebre entre los exones 5-11 de 8,3 kb, y en su estructura se ha identificado m·ltiples dominios proteicos: ganchos AT, un dominio de ADN metiltransferasa (transcriptional repression domain, TRD), un dominio PHD (Plant homology domain), un dominio activador de la transcripci¾n (TA) y un dominio SET o Su (var)3-9, enchancer-of-zeste, trithorax (7) (Figura 1).
Figura 1. Estructura de la proteĒna MLL.
Las alteraciones en el gen MLL incluyen deleciones, duplicaciones, inversiones y translocaciones recĒprocas; en estas ·ltimas se generan proteĒnas de fusi¾n por la interacci¾n con otros genes, denominados fusion partner genes (FPGs), reemplazando asĒ los dominios de represi¾n transcripcional y de se±alizaci¾n nuclear ubicados en el extremo N-terminal de la proteĒna MLL, por los extremos C-terminales de los genes de fusi¾n. Los puntos de quiebre que se observan en las leucemias de novo se concentran en la regi¾n centromķrica, mientras que en las leucemias del lactante y secundarias a tratamiento lo hacen en la regi¾n telomķrica (5-7). Los genes de fusi¾n mßs frecuentes son: 9p 21-22 (t 9;11) en MLL-AF9, 4q21 (t 4;11) en MLL-AF4 y 19p13 (t 11;19) en MLL-ENL. MLL-AF4 se ve con mayor frecuencia en LLA, MLL-AF9 es mßs frecuente en LMA y MLL-ENL es com·n en ambos tipos de leucemia (8) (Figura 2).
Figura 2. Genes de fusi¾n con MLL.
Se ha postulado que estas proteĒnas de fusi¾n participan en la leucemogķnesis mediante el aumento de la expresi¾n del gen HOX, que codifica para factores transcripcionales, mecanismo que serĒa crĒtico para las leucemias relacionadas con el gen MLL. Otro mecanismo descrito para estas proteĒnas de fusi¾n es el aumento de la expresi¾n de la tirosin kinasa FLT3, como otro paso para la inducci¾n de leucemia del gen MLL (4-6).
En la actualidad se dispone de m·ltiples tķcnicas moleculares para evaluar la expresi¾n del perfil genķtico del gen MLL, entre ellas:
La leucemia linfoblßstica en ni±os ocurre con mayor frecuencia entre los 2 y 5 a±os. Existen diversos factores de mal pron¾stico, como hiperleucocitosis en el momento del diagn¾stico, edad de inicio menor de 1 a±o o mayor de 10 a±os, portaci¾n de citogenķtica adversa (t 9;22 o cromosoma Philadelphia, hipodiploidĒa, etc.) y mala respuesta a tratamiento inicial. Esto ha permitido clasificar a los pacientes y programar un tratamiento estratificado, con lo que la sobrevida a 5 a±os alcanza hasta 90% en los ni±os mayores, no asĒ en el grupo de lactantes, donde la sobrevida a los 4 a±os llega s¾lo a 55%. Estas diferencias tan marcadas no se explican s¾lo por los tratamientos disponibles, sino por la biologĒa misma de la enfermedad (3-7).
La LLA en lactantes corresponde a una forma mßs inmadura de diferenciaci¾n, como lo demuestra la expresi¾n de antĒgenos de superficie: CD 10 (-), CD19 (+), HLA DR (+) y ademßs coexpresan antĒgenos mieloides. En 75% de los casos en lactantes se encuentran alteraciones del gen MLL, que constituyen un factor de peor pron¾stico de la enfermedad por sĒ mismas y se asocian a hiperleucocitosis y a menor edad de presentaci¾n, con tasas de sobrevida a 5 a±os de 35 a 50%. Dentro de los rearreglos MLL, la translocaci¾n 4;11 (MLL-AF4) es la mßs frecuente y la que se asocia a menor tasa de sobrevida en este grupo etario, cercana a 15% a 5 a±os de seguimiento. Otras alteraciones estructurales del gen 11q23, como su deleci¾n, tambiķn se asocian a caracterĒsticas clĒnicas de peor pron¾stico (3-5).
Entre los mecanismos planteados para explicar la patogenia de la leucemia aguda en el perĒodo de lactante, las translocaciones del gen MLL tienen un papel importante. Diversos estudios en gemelos que han desarrollado leucemia han llevado a plantear que las cķlulas neoplßsicas de este tipo de leucemia son de origen fetal y sufren transformaciones genķticas secundarias in ·tero y en la vida postnatal, resultando en alteraciones del 11q23. Esto origina el ōfenotipo mutadorö, que favorece la rßpida acumulaci¾n de mutaciones complementarias y deteriora el reconocimiento del quiebre de la doble banda de ADN y los checkpoints del ciclo celular. Este modelo se diferencia de los propuestos para otras neoplasias, en los cuales se necesita al menos dos mutaciones complementarias consecutivas que afecten una, a la diferenciaci¾n celular y otra, al control de proliferaci¾n y apoptosis celular (4-7).
Estudios in vitro han demostrado que las cķlulas leucķmicas de LLA en lactantes son mßs resistentes al tratamiento con corticoides y L-asparraginasa que otras cķlulas leucķmicas, pero son mßs sensibles a la citarabina. Entre los mecanismos de resistencia asociados a la portaci¾n de translocaciones del gen MLL estß la inhibici¾n de la actividad transcripcional del gen p53 mediante la down-regulation de la inducci¾n de los genes p21, mdm2 y Bax en respuesta al da±o del ADN (5-7).
La leucemia mieloide aguda corresponde a 15 a 20% de las leucemias agudas de la edad pedißtrica y es ligeramente mßs frecuente en lactantes que en ni±os mayores. A diferencia de la LLA, la LMA pedißtrica tiene un comportamiento similar a la del adulto y en ella la portaci¾n de rearreglos del gen 11q23 no empeora el pron¾stico (5). Diversos estudios han demostrado tasas de portaci¾n de rearreglos de MLL hasta en 35% de los casos de LMA del lactante, con sobrevidas cercanas a 50% a 4 a±os de seguimiento. En este tipo de leucemia la translocaci¾n 9;11 (MLL-AF9) es la mßs frecuente y se encuentra en cerca de la mitad de los casos. Tambiķn se ha descrito la fuerte asociaci¾n entre las alteraciones de MLL y los fenotipos LMA M4 y M5 (4, 5).
En distintas series de LMA en lactantes el factor edad fue determinante para la sobrevida libre de enfermedad y las tasas de recaĒdas, dado que este grupo etario presenta menos resistencia a los tratamientos quimioterapķuticos, pero tiene mayor mortalidad asociada a terapia (5).
Los tratamientos quimioterapķuticos con citot¾xicos pueden tener como efecto colateral el desarrollo de neoplasias secundarias, tanto precoces, dentro de los tres primeros a±os de finalizado el tratamiento como tardĒas, que se presentan siete o mßs a±os despuķs de completar el tratamiento (3, 4).
Se ha descrito el desarrollo de leucemia mieloide aguda en pacientes previamente tratados por leucemias o tumores s¾lidos con inhibidores de la Topoisomerasa II, como epipodofilotoxinas (etop¾sido, tenop¾sido), antraciclinas (adriamicina, daunorrubina) y derivados de la dioxipiperazina. En estos pacientes se ha encontrado una incidencia entre 70 a 90% de alteraciones en el 11q23, especialmente amplificaciones y translocaciones, siendo ķstas las mßs frecuentes en los pacientes pedißtricos. En los casos de LMA precoz se encuentran translocaciones balanceadas del gen MLL, en cambio en las LMA tardĒas se detectan translocaciones desbalanceadas, especialmente en pacientes con antecedente de sĒndromes mielodisplßsicos. Las translocaciones (9;11) y (11;19) son las mßs comunes, principalmente en pacientes con antecedente de uso de epipodofilotoxinas. Este grupo presenta tasas de remisi¾n similares a las LMA de novo, pero la sobrevida final es baja a pesar de quimioterapia o transplante de precursores hematopoyķticos (3, 5). La LLA secundaria a tratamiento es menos frecuente que la LMA y se describe con el uso previo de agentes alquilantes (ciclofosfamida, cisplatino) y radiaci¾n. En estos casos la translocaci¾n (4;11) es la mßs frecuente, con resultados de sobrevida a largo plazo limitados (3, 4).
El anßlisis del gen MLL y sus alteraciones ha permitido el avance del conocimiento de la patogenia de la leucemia aguda en pediatrĒa, patologĒa que por su frecuencia y tasa de mortalidad motiva al mundo cientĒfico en la b·squeda y desarrollo de mejores parßmetros para caracterizar a los pacientes portadores y optimizar los beneficios y reducir los efectos secundarios de los tratamientos antineoplßsicos.
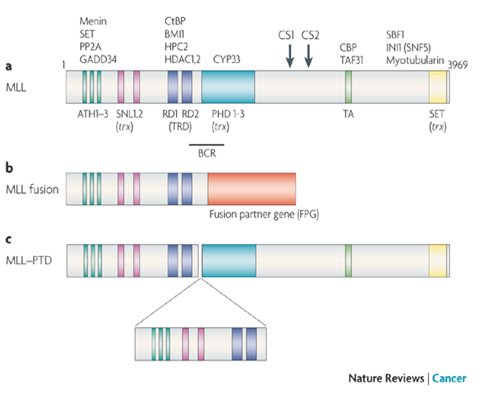 Figura 1. Estructura de la proteĒna MLL.
Figura 1. Estructura de la proteĒna MLL.
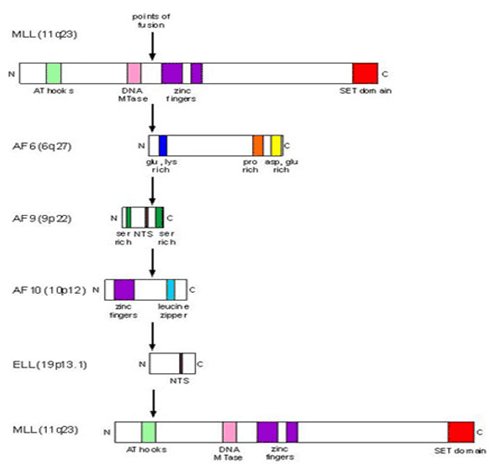 Figura 2. Genes de fusi¾n con MLL.
Figura 2. Genes de fusi¾n con MLL.
 Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Este texto corresponde a un trabajo de revisi¾n preparado por sus autores en el desarrollo del Curso y Seminarios de OncologĒa Bßsica, realizado por el Centro de OncologĒa Preventiva y la Escuela de Postgrado de la Facultad de Medicina de la Universidad de Chile entre abril y agosto de 2008. El Director del Curso es el Dr. Josķ Manuel Ojeda.
Autores:
Fernando Bracho Milic[1], Ximena Claverie Ramos[1]
Citaci¾n: Bracho F, Claverie X. MLL gene and leukemia in children. Medwave 2009 Abr;9(4):e3855 doi: 10.5867/medwave.2009.04.3855
Fecha de publicaci¾n: 1/4/2009
Nos complace que usted tenga interķs en comentar uno de nuestros artĒculos. Su comentario serß publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la direcci¾n editorial considera que su comentario es: ofensivo en alg·n sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas polĒticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisi¾n por pares.
A·n no hay comentarios en este artĒculo.
Para comentar debe iniciar sesi¾n
Medwave publica las vistas HTML y descargas PDF por artĒculo, junto con otras mķtricas de redes sociales.
Hilden JM, Smith FO, Frestedt JL, McGlennen R, Howells WB, Sorensen PH, et al. MLL gene rearrangement, cytogenetic 11q23 abnormalities, and expression of the NG2 molecule in infant acute myeloid leukemia. Blood. 1997 May 15;89(10):3801-5. | PubMed |Behm FG, Raimondi SC, Frestedt JL, Liu Q, Crist WM, Downing JR, et al. Rearrangement of the MLL gene confers a poor prognosis in childhood acute lymphoblastic leukemia, regardless of presenting age. Blood. 1996 Apr 1;87(7):2870-7. | PubMed |Aplan PD. Chromosomal translocations involving the MLL gene: molecular mechanisms. DNA Repair (Amst). 2006 Sep 8;5(9-10):1265-72. Epub 2006 Jun 21. | CrossRef | PubMed | PMC |Armstrong SA, Golub TR, Korsmeyer SJ. MLL-rearranged leukemias: insights from gene expression profiling. Semin Hematol. 2003 Oct;40(4):268-73. | CrossRef | PubMed |Chowdhury T, Brady HJ. Insights from clinical studies into the role of the MLL gene in infant and childhood leukemia. Blood Cells Mol Dis. 2008 Mar-Apr;40(2):192-9. Epub 2007 Oct 1. | CrossRef | PubMed |Wiederschain D, Kawai H, Shilatifard A, Yuan ZM. Multiple mixed lineage leukemia (MLL) fusion proteins suppress p53-mediated response to DNA damage. J Biol Chem. 2005 Jul 1;280(26):24315-21. Epub 2005 Apr 25. | CrossRef | PubMed |






 Estudios originales
Estudios originales