Revista Biomķdica Revisada Por Pares

 Para Descargar PDF debe Abrir sesi¾n.
Para Descargar PDF debe Abrir sesi¾n.
Este texto completo es una transcripci¾n editada de la conferencia que se dict¾ en el XLVIII Congreso Chileno de PediatrĒa realizado en Vi±a del Mar entre el 26 y el 29 de Noviembre de 2008. El congreso fue organizado por la Sociedad Chilena de PediatrĒa bajo la presidencia de la Dra. Lidya TellerĒas C.
La enfermedad de Kawasaki (EK) tiene una presentaci¾n clĒnica compleja y no se ha logrado demostrar su etiologĒa, por lo tanto no se sabe c¾mo prevenirla; sin embargo existe un tratamiento efectivo.
Kawasaki describi¾ por primera vez la enfermedad que lleva su nombre en 1967, como un sĒndrome febril ¾culo-oro-cutßneo, acrodescamativo, con descamaci¾n de la piel alrededor de u±as, palmas y plantas, con o sin linfadenitis cervical aguda no supurativa en 50 lactantes y ni±os. Todos los pacientes del estudio tenĒan fiebre mayor de 38░C de al menos 6 dĒas duraci¾n, a pesar de recibir antibi¾ticos; en 98% de ellos se constat¾ inyecci¾n conjuntival sin secreci¾n, signo que hoy se considera como el pilar del diagn¾stico clĒnico de la enfermedad; 86% present¾ rash o eritema palmoplantar sin vesĒculas ni bulas; 96% tenĒa labios rojos, secos, erosionados y figurados, enantema sin vesĒculas ni aftas y, en forma ocasional, lengua de ōfresaö, lo que liga a la enfermedad con la posibilidad etiol¾gica de Streptococcus pyogenes; 66% de los pacientes desarroll¾ linfadenitis cervical aguda no supurativa; 44%, edema de manos y pies en lactantes y preescolares; y 98% tuvo acrodescamaci¾n, que a partir de la segunda semana, pasada la etapa aguda de la enfermedad, se limit¾ s¾lo a manos y pies. El rango de edad de los pacientes fue de 2 meses a 9 a±os y 1 mes; 54% eran menores de dos a±os. El sĒndrome cur¾ espontßneamente, sin secuelas, recurrencias ni contagiosidad (1).
La incidencia de EK en Jap¾n ha aumentado en forma progresiva desde 1970, posiblemente debido a mejor registro de la patologĒa, llegando a 187 por 100.000 ni±os menores de 4 a±os en 2007 y 132 por 100.000 en menores de 5 a±os. Esta cifra es muy superior a la de Estados Unidos y Europa, donde la incidencia es 3 a 10 por 100.000 ni±os menores de 5 a±os. La mortalidad oscila entre 0,1 y 2%. La EK es mßs frecuente en varones, con una relaci¾n de 1,6:1 y 90% de los casos ocurre en menores de 10 a±os, aunque es poco frecuente en los menores de 6 meses en quienes la presentaci¾n clĒnica es incompleta (Kawasaki atĒpico) y tiene peor pron¾stico. Los ni±os mayores de 8 a±os tienen mayor riesgo de desarrollar cardiopatĒa (2).
Marian Melish fue la primera en describir la enfermedad fuera de Jap¾n, especĒficamente en Hawaii, en un estudio en que describi¾ n·mero de pacientes y porcentaje acumulativo de casos seg·n edad en ni±os menores de 18 a±os (Fig. 1).
Este estudio demostr¾ la importancia del factor ambiental, ya que la incidencia en ni±os de origen japonķs residentes en Hawaii, quienes poseen un estilo de vida occidental, fue superior a la de los ni±os japoneses y lo mismo ocurre con ni±os caucßsicos residentes de Hawai, en comparaci¾n a aquellos que habitan en el continente (3) (Tabla I).
Tabla I. Incidencia de enfermedad de Kawasaki en ni±os seg·n origen.
No estß claro si la causa de la EK es un agente ·nico, viral o bacteriano, agentes m·ltiples con una patogenia com·n o un superantĒgeno asociado a un agente; lo que sĒ estß claro es que el resultado final es una marcada activaci¾n del sistema inmune.
Trang public¾ un estudio en un modelo de ratas en el que se inyect¾ a estos animales un extracto de pared celular de Lactobacillus casei (EPCLC), despuķs de lo cual se observ¾ la aparici¾n de arteritis y aneurismas coronarios. La respuesta al EPCLC re·ne todas las caracterĒsticas de la respuesta mediada por superantĒgenos y lo mßs relevante fue que la presencia de actividad superantigķnica se correlacion¾ directamente con la inducci¾n de arteritis coronaria (4).
Rowley investig¾ el origen de la enfermedad en un anßlisis de varios estudios que describen la presencia de cuerpos de inclusi¾n citoplasmßtica asociados a m·ltiples agentes que desencadenan distintas patogenias (5). Para la mayor parte de estos agentes no existe evidencia adecuada o bien, falta confirmaci¾n. Actualmente estß en investigaci¾n la hip¾tesis de la existencia de un agente viral ARN persistente, previamente no reconocido, que infecta a las cķlulas y desata la respuesta inmune induciendo la formaci¾n de cuerpos de inclusi¾n citoplasmßtica persistentes (Tabla II).
La formaci¾n de aneurismas se explicarĒa por el paso del agente desde el espacio intravascular al extravascular en el interior de un monocito o macr¾fago, dando inicio a un fen¾meno inflamatorio con agregaci¾n plaquetaria y liberaci¾n de metaloproteinasas de matriz que alteran la lßmina elßstica interna y externa, lo que provoca la desestructuraci¾n de la pared vascular con formaci¾n posterior del aneurisma (Fig. 2).
Figura 2. Eventos que causan los aneurismas coronarios en la enfermedad de Kawasaki.
La hip¾tesis sobre la patogenia de la EK postula que el agente ingresa por la vĒa respiratoria y penetra a travķs del epitelio bronquial, donde es captado por los macr¾fagos tisulares lo que inicia la respuesta inmune innata. Posteriormente el antĒgeno se transporta hacia los n¾dulos linfßticos locales, donde desencadenan la respuesta inmune adaptativa. Ademßs estos macr¾fagos pasan al sistema circulatorio y se dirigen a travķs de los vasos sanguĒneos hacia distintos ¾rganos como pßncreas, glßndulas salivales, pr¾stata y otros. En el epitelio bronquial el agente desencadena la producci¾n de proteĒnas virales que se engloban dentro de cuerpos de inclusi¾n citoplasmßtica que no son reconocidos por el sistema inmune, por lo que pueden permanecer en forma persistente (Fig. 3).
Figura 3. Patogenia propuesta para la enfermedad de Kawasaki.
La patologĒa de la EK se caracteriza por un compromiso de las arterias medianas, con inflamaci¾n de la t·nica Ēntima y de la zona perivascular que cursa en seis etapas, cuyas caracterĒsticas se describen a continuaci¾n.
La disociaci¾n de la capa media debido al edema ocurre entre 6 y 8 dĒas despuķs del inicio de la enfermedad; el compromiso completo de la pared ocurre a los 10 dĒas; el da±o severo y el aneurisma se presentan a los 12 dĒas de evoluci¾n, la inflamaci¾n persiste hasta los 25 dĒas y las cķlulas inflamatorias desaparecen alrededor de los 40 dĒas.
Los ¾rganos afectados por la enfermedad y la correspondiente alteraci¾n de ķstos se describe a continuaci¾n:
Se ha afirmado que la EK es la poliarteritis nodosa infantil (PNI); sin embargo los estudios anatomopatol¾gicos han demostrado que son entidades distintas, porque el compromiso de la EK ocurre antes de que los vasos ingresen a los ¾rganos, tiene una evoluci¾n sincr¾nica con las etapas de la enfermedad y tiene predominio de macr¾fagos y monocitos en la fase aguda, ademßs de edema; en cambio la PNI ocurre antes y despuķs del ingreso vascular a los ¾rganos, tiene una evoluci¾n clĒnica asincr¾nica y en la histologĒa predominan los neutr¾filos y la necrosis fibrinoide (6).
El patr¾n clĒnico consistente con EK incluye fiebre alta con alzas bruscas que dura mßs de 5 dĒas; desarrollo de eritema y edema palmoplantar, con descamaci¾n del lecho ungueal de dedos y ortejos a partir de la segunda a tercera semana desde el inicio; inyecci¾n no purulenta de la conjuntiva bulbar; exantema maculopapular escarlatiniforme o multiforme precoz y de duraci¾n breve, menor de 5 dĒas; compromiso perianal con descamaci¾n; presencia de fisura labial, eritema de la mucosa oral y lengua con aspecto de ōfresaö; adenopatĒa cervical unilateral de mßs de 1,5 cm de dißmetro; y otras manifestaciones como irritabilidad extrema, sĒntomas cardiovasculares, artritis, artralgias, diarrea, disfunci¾n hepßtica, meningitis asķptica, leucocituria y uretritis.
No son signos compatibles con EK: fiebre moderada o baja con resoluci¾n espontßnea en menos de 5 dĒas; descamaci¾n palmoplantar precoz; conjuntivitis purulenta; exantema tardĒo o de mßs de 5 dĒas de evoluci¾n; faringitis sin otras alteraciones de la cavidad oral; adenopatĒas difusas y paciente en buen estado general. En presencia de estos signos el diagn¾stico de EK es dudoso.
Otras pistas clĒnicas que apoyan el diagn¾stico son: eritema e induraci¾n de la cicatriz de BCG, rash o descamaci¾n perineal, hidrops vesicular, uveĒtis, hipertensi¾n, meningitis asķptica, poliartritis, enfermedad vascular perifķrica manifestada como pulso alterado o isquemia y gangrena de extremidades. Asimismo se puede encontrar aumento de PCR y VHS, anemia moderada, leucocitosis y desviaci¾n a izquierda, trombocitosis, piuria asķptica, hiponatremia, aumento de transaminasas y gama glutamiltransferasa, hipoalbuminemia, alteraci¾n de lĒpidos plasmßticos, hipereosinofilia mßs IgE sķrica aumentada e hiperrefringencia de las coronarias.
La EK incompleta es una variante de la enfermedad en la cual el paciente no re·ne todos los criterios clĒnicos, por lo tanto se trata de un caso incompleto o atĒpico que a menudo se diagnostica en forma tardĒa. Esta forma es mßs frecuente en el primer a±o de vida, especialmente en menores de seis meses y los lactantes son mßs propensos a desarrollar aneurismas coronarios; de hecho la incidencia de aneurismas coronarios en pacientes que no re·nen los criterios diagn¾sticos alcanza a 25%. Se debe considerar el diagn¾stico en todo ni±o con fiebre de mßs de cinco dĒas de evoluci¾n sin origen explicable, mßs la presencia de alg·n criterio adicional. Por ello es fundamental solicitar ecocardiografĒa en casos de fiebre inexplicable y evidencias de inflamaci¾n en los exßmenes de laboratorio (VHS y PCR). Seg·n la investigaci¾n realizada por Anderson el retardo en el diagn¾stico no se asocia con el tipo de profesional mķdico que atiende al paciente, el n·mero de antimicrobianos utilizados o el n·mero de visitas. Los pacientes con diagn¾stico tardĒo presentan los signos clßsicos, pero de comienzo disperso en el tiempo y desarrollan mayor cantidad de aneurismas que los que tienen diagn¾stico precoz; por ello es necesario educar para sospechar la enfermedad en ni±os con fiebre y/o rash (7).
Newburger estableci¾ en 1986 la efectividad de la inmunoglobulina intravenosa (IgIV) en un estudio controlado, aleatorio y multicķntrico en el cual administr¾ IgIV en dosis de 400 mg/kg/dĒa por un perĒodo de cuatro dĒas, asociada a Aspirina« en dosis de 80 a 100 mg/kg/dĒa durante 14 dĒas, a un grupo de pacientes; y s¾lo Aspirina« a otro grupo. En ambos casos se inici¾ el tratamiento antes del dķcimo dĒa desde el comienzo de la fiebre. En el grupo que se trat¾ s¾lo con Aspirina« 28% de los pacientes desarroll¾ anomalĒas coronarias, dilataci¾n y aneurismas; en cambio, los pacientes que recibieron IgIV presentaron estas anomalĒas en s¾lo 8% de los casos (8).
En 1991 el mismo autor determin¾ que el mejor efecto se obtiene con una dosis ·nica de 2g/kg de IgIV, que es la indicaci¾n que prevalece en la actualidad, gracias a un estudio en que compar¾ esta dosis con una dosis mßs baja de 400mg/kg/dĒa por un perĒodo de cuatro dĒas, ademßs de administraci¾n de Aspirina« en ambos grupos. La incidencia de anomalĒas coronarias fue inferior con la dosis ·nica (4,6% de los casos) que con la dosis reiterada de 400 mg (9,1% de los casos). Ademßs los marcadores de inflamaci¾n, como alfa-1 antitripsina y proteĒna C reactiva redujeron su valor en forma mßs precoz con la dosis ·nica (9).
No se conoce con exactitud el mecanismo de acci¾n de la IgIV, pero se recomienda administrar este tratamiento entre el quinto y el dķcimo dĒa de evoluci¾n aguda, puesto que antes de este plazo no previene alteraciones coronarias. Por otra parte se recomienda emplear tal esquema despuķs del dķcimo dĒa en ni±os con evidencia de inflamaci¾n sistķmica, fiebre persistente y aneurismas coronarios. Alrededor de 5% de los ni±os desarrollan aneurismas a pesar del tratamiento y otro 1% presenta aneurismas gigantes. Respecto a la Aspirina«, se sabe que el uso de ķsta en dosis altas, 80 a 100 mg/kg/dĒa junto a la IgIV tiene un efecto antiinflamatorio aditivo. El esquema se mantiene hasta que el paciente permanezca afebril durante 48 a 72 horas y posteriormente se utilizan dosis bajas hasta 6 a 8 semanas tras el inicio de la enfermedad o bien, en forma indefinida en caso de que existan alteraciones coronarias. La Aspirina« no previene el desarrollo de aneurismas aunque se utilice en dosis elevadas.
Se habla de fracaso del tratamiento cuando la fiebre persiste por 36 horas o mßs tras la administraci¾n de IgIV. La incidencia de fracaso de este tratamiento es 10%.
La recomendaci¾n actual es aplicar una segunda dosis de Ig de 2 g/kg antes de 10 dĒas de evoluci¾n aunque exista respuesta a la dosis inicial, porque ademßs de inducir respuesta en los casos de fracaso al tratamiento inicial esta medida reduce la prevalencia de lesiones coronarias (10). En caso de aneurismas gigantes en fase aguda o subaguda se puede agregar al tratamiento descrito Abciximab, un inhibidor del receptor de la glicoproteĒna plaquetaria IIb/IIIa, ya que esta medida mejora el pron¾stico a largo plazo debido a que disminuye el dißmetro de los aneurismas. La adici¾n de metilprednisolona en dosis de 30 mg/kg por vĒa endovenosa a la terapia con IgIV y Aspirina« dentro de los primeros 9 dĒas de evoluci¾n reduce el tiempo de hospitalizaci¾n y los parßmetros inflamatorios, sin embargo no presenta diferencias significativas en la reducci¾n de anomalĒas coronarias y de todos modos se requiere una segunda dosis de IgIV. Newburger, en un estudio aleatorio, doble ciego y controlado en el que participaron ocho centros con un total de 195 pacientes con EK, concluy¾ que no hay evidencia suficiente que apoye el uso de pulsos de metilprednisolona junto al tratamiento primario convencional con IgIV (11).
La EK es una enfermedad autolimitada que cursa con inflamaci¾n del sistema vascular que normalmente se resuelve en dos a tres meses. La mayorĒa de los pacientes estßn sanos antes de la enfermedad y se recuperan en forma permanente desde el punto de vista funcional, sin secuelas aparentes. El pron¾stico a largo plazo (20 a 30 a±os) depende del estado coronario uno o dos meses desde el comienzo de la enfermedad. La proporci¾n de ni±os que se recuperan tras recibir el tratamiento con IgIV es superior a 95%; la mayorĒa tienen un estado inmunol¾gico normal y s¾lo manifiestan problemas relacionados con el da±o coronario.
El compromiso coronario presenta un espectro que varĒa desde la sobrevida hasta la muerte y puede presentar diversas manifestaciones: ausencia de alteraciones en la ecocardiografĒa; dilataci¾n coronaria de corta evoluci¾n que se resuelve en 2 a 6 meses de evoluci¾n; desarrollo de aneurismas medianos, de menos de 7 mm de dißmetro; casos con aneurismas gigantes, que miden mßs de 8 mm; y por ·ltimo, en menor proporci¾n, muerte precoz durante el curso de la enfermedad.
Los pacientes sin alteraci¾n en las ecocardiografĒas no tienen mayor riesgo de muerte o problemas cardĒacos en los siguientes 20 a 30 a±os, aunque se demuestre la presencia de cambios microsc¾picos. Por tal raz¾n la recomendaci¾n es suspender la dosis bajas de Aspirina« a los dos o tres meses, cuando los parßmetros inflamatorios (VHS, PCR y plaquetas) se normalicen; y educar en estilos de vida saludable con ķnfasis en la prevenci¾n del da±o cardiovascular o aterosclerosis. Con esto no es necesario hacer control cardiol¾gico entre los dos meses y el a±o de edad. En los mayores de dos a±os es necesario controlar con perfil lipĒdico.
Los pacientes con dilataci¾n coronaria inicial que revierte en 2 a 6 meses tampoco tienen mayor tasa de mortalidad en los siguientes 20 a 30 a±os; en ella se recomienda administrar dosis bajas de Aspirina« desde el a±o de edad en forma permanente, ademßs de hacer ķnfasis en la vida saludable y el control cardiovascular. Se debe repetir la ecocardiografĒa entre los seis meses y el a±o de evoluci¾n hasta obtener al menos dos ecocardiografĒas normales. El control cardiol¾gico es opcional.
Los aneurismas medianos se resuelven dentro de los primeros dos a±os en 50% de los casos, pero las ßreas afectadas son anormales, no se dilatan con el ejercicio ni progresan a estenosis en los 20 a±os siguientes, lo que otorga a la patologĒa un diagn¾stico reservado. En 50% de los casos estas alteraciones persisten lo cual depende en parte de su localizaci¾n, ya que son mßs frecuentes en determinadas arterias coronarias, formas y tama±os; en este caso pueden progresar a estenosis. Por lo tanto, en caso de aneurismas medianos se recomienda la administraci¾n de dosis bajas de Aspirina« en forma indefinida, asociada a drogas antiplaquetarias como clopidogrel o persantina, ademßs de educaci¾n en estilo de vida saludable y control cardiovascular.
Los aneurismas gigantes se asocian a riesgo de infarto durante los primeros dos a±os de evoluci¾n. El riesgo de estenosis depende del tama±o, forma y localizaci¾n del aneurisma, no obstante el riesgo de ruptura aneurismßtica es bajo y parece estar limitado a los primeros dos a±os. Por lo tanto, en esta patologĒa se debe aplicar una baterĒa de estudios que incluye ecocardiografĒa, ECG, holter de arritmias, angiografĒa, test de perfusi¾n con tecnecio, resonancia magnķtica, tomografĒa ultrarrßpida y ultrasonido intravascular. El manejo consiste en mantener una adecuada anticoagulaci¾n con warfarina mßs un antiagregante plaquetario, sea Aspirina« o clopidogrel. Se debe realizar estudios imagenol¾gicos en forma peri¾dica con test de ejercicio y drogas, ademßs de SPECT. Se debe efectuar control cardiol¾gico regular y enfatizar en estilo de vida saludable. Se podrĒa requerir tratamientos intervencionales, como plasmin¾geno tisular, angioplastĒa con bal¾n en caso de estenosis secundaria de las coronarias, instalaci¾n de stent, tķcnicas de ablaci¾n de trombos con rototblader, bypass coronarios en casos de estenosis severa y trasplante cardĒaco.
El pron¾stico de la EK ha mejorado en los ·ltimos diez a±os en forma concomitante con la expectativa de vida, gracias al apoyo cardiol¾gico y a la mayor disponibilidad de tratamientos. Es importante derivar a estos ni±os a profesionales expertos y mantener una conexi¾n estrecha con ellos; aquellos con problemas graves e inusuales deben ser derivados rßpidamente.
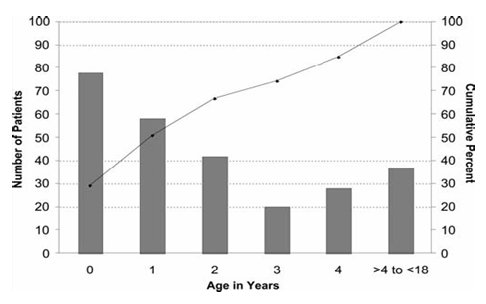 Figura 1. N·mero y porcentaje acumulativo seg·n edad en ni±os menores de 18 a±os con enfermedad de Kawasaki, Hawai 1996-2001.
Figura 1. N·mero y porcentaje acumulativo seg·n edad en ni±os menores de 18 a±os con enfermedad de Kawasaki, Hawai 1996-2001.
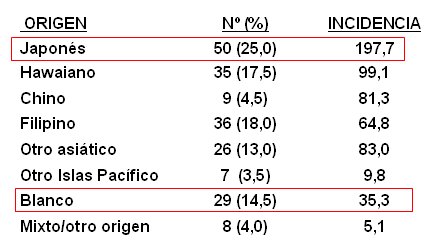 Tabla I. Incidencia de enfermedad de Kawasaki en ni±os seg·n origen.
Tabla I. Incidencia de enfermedad de Kawasaki en ni±os seg·n origen.
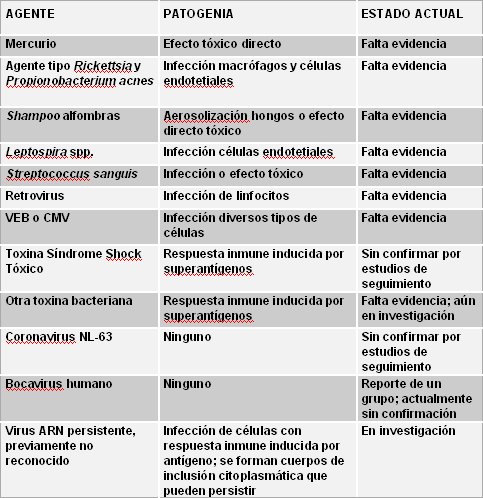 Tabla II. B·squeda de la causa de la enfermedad de Kawasaki en cuerpos de inclusi¾n citoplasmßtica causados por m·ltiples agentes.
Tabla II. B·squeda de la causa de la enfermedad de Kawasaki en cuerpos de inclusi¾n citoplasmßtica causados por m·ltiples agentes.
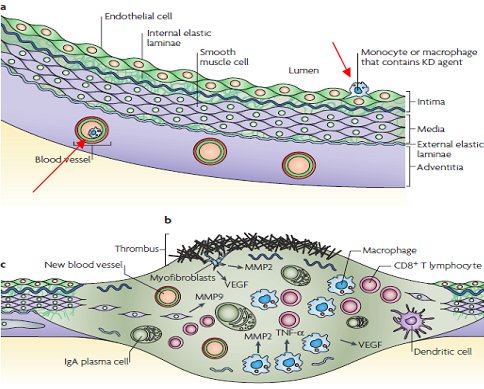 Figura 2. Eventos que causan los aneurismas coronarios en la enfermedad de Kawasaki.
Figura 2. Eventos que causan los aneurismas coronarios en la enfermedad de Kawasaki.
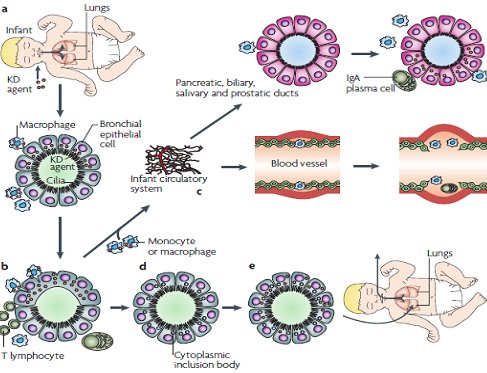 Figura 3. Patogenia propuesta para la enfermedad de Kawasaki.
Figura 3. Patogenia propuesta para la enfermedad de Kawasaki.
 Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Este texto completo es una transcripci¾n editada de la conferencia que se dict¾ en el XLVIII Congreso Chileno de PediatrĒa realizado en Vi±a del Mar entre el 26 y el 29 de Noviembre de 2008. El congreso fue organizado por la Sociedad Chilena de PediatrĒa bajo la presidencia de la Dra. Lidya TellerĒas C.
Autor:
Antonio Banfi[1]
Citaci¾n: Banfi A. Update on Kawasaki disease. Medwave 2009 Sep;9(9):e4153 doi: 10.5867/medwave.2009.09.4153
Fecha de publicaci¾n: 1/9/2009
Nos complace que usted tenga interķs en comentar uno de nuestros artĒculos. Su comentario serß publicado inmediatamente. No obstante, Medwave se reserva el derecho a eliminarlo posteriormente si la direcci¾n editorial considera que su comentario es: ofensivo en alg·n sentido, irrelevante, trivial, contiene errores de lenguaje, contiene arengas polĒticas, obedece a fines comerciales, contiene datos de alguna persona en particular, o sugiere cambios en el manejo de pacientes que no hayan sido publicados previamente en alguna revista con revisi¾n por pares.
A·n no hay comentarios en este artĒculo.
Para comentar debe iniciar sesi¾n
Medwave publica las vistas HTML y descargas PDF por artĒculo, junto con otras mķtricas de redes sociales.
Kawasaki T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Arerugi. 1967 Mar;16(3):178-222. | PubMed |Nakamura Y. Forty one years after first report of Kawasaki Disease in Japan; 2008: Taiwan, IX International Symposium of Kawasaki Disease. | Link |Holman RC, Curns AT, Belay ED, Steiner CA, Effler PV, Yorita KL, et al. Kawasaki syndrome in Hawaii. Pediatr Infect Dis J. 2005 May;24(5):429-33. | CrossRef | PubMed |Duong TT, Silverman ED, Bissessar MV, Yeung RS. Superantigenic activity is responsible for induction of coronary arteritis in mice: an animal model of Kawasaki disease. Int Immunol. 2003 Jan;15(1):79-89. | CrossRef | PubMed |Rowley AH, Baker SC, Orenstein JM, Shulman ST. Searching for the cause of Kawasaki disease--cytoplasmic inclusion bodies provide new insight. Nat Rev Microbiol. 2008 May;6(5):394-401. | CrossRef | PubMed |Takahashi K, Oharaseki T, Yokouchi Y, Naoe S. Pathological characteristics of systemic vascular lesions in Kawasaki disease and other vasculitis syndrome; 2008: Taiwan, IX International Symposium of Kawasaki Disease. | Link |Anderson MS, Todd JK, Glodķ MP. Delayed diagnosis of Kawasaki syndrome: an analysis of the problem. Pediatrics. 2005 Apr;115(4):e428-33. | CrossRef | PubMed |Newburger JW, Takahashi M, Burns JC, Beiser AS, Chung KJ, Duffy CE, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. N Engl J Med. 1986 Aug 7;315(6):341-7. | CrossRef | PubMed |Newburger JW, Takahashi M, Beiser AS, Burns JC, Bastian J, Chung KJ, et al. A single intravenous infusion of gamma globulin as compared with four infusions in the treatment of acute Kawasaki syndrome. N Engl J Med. 1991 Jun 6;324(23):1633-9. | CrossRef | PubMed |






 Estudios originales
Estudios originales