
Key Words: Huntington disease; inflammation; nervous system; neurodegeneration; periodontitis
Abstract
Huntington's disease is a neurodegenerative disorder caused by the expansion of a CAG triplet in the huntingtin gene. It presents with physical, cognitive and psychiatric impairment at different ages in the adult, and has a fatal prognosis. Other than the number of triplet repetitions, there seem to be other factors that explain the onset of this disease at an earlier age. It is well known that neuroinflammation has a key role in neurodegenerative disorders; Huntington's disease is not an exception to that rule. Neuroinflammation exacerbates neuronal damage produced by mutation, by initiating aberrant activation of microglia cell, as well as astrocyte and dendritic cell dysfunction; also compromising the blood-brain barrier and activating the complement cascade. The latter as a direct and indirect effect of the mutation and other stimuli such as chronic infections. In this study, periodontitis is presented as a model of chronic oral infection and a systemic inflammation source. We hypothesize the potential role of periodontitis in Huntington's disease, and the mechanisms by which it contributes to the early onset and progress of the disease. We considered experimental studies, systematic reviews, meta-analyses, published in both Spanish and English, obtained from the PubMed and SciELO databases. There are various mechanisms that generate brain inflammation in these patients; mechanisms of innate immunity being especially prominent. Chronic oral-dental infections, such as periodontal disease, may be an exacerbating factor that adds to the neuroinflammation of Huntington’s disease.
Introduction
Huntington's disease is a neurodegenerative disorder that affects primarily adults. It is inherited as an autosomal dominant disease manifested with physical, cognitive and mental deterioration, which is eventually fatal [1].The main cause of this syndrome is a dynamic mutation based on a specific repetitive sequence of cytosine, adenine, guanine in exon 1 of the huntingtin gene. The alleles (CAG)40 and more repetitions have a high penetrance and cause Huntington's disease. There is an inverse relationship between the size of the repetition and the age of onset [2]. However, this would not be the only factor in the youth forms of Huntington's disease. Recent studies on immune system and Huntington's disease [1] show that neuronal damage does not only depend on the toxicity of mutant huntingtin and its interactions, but also on cerebral environment and inflammatory conditions [3],[4]. Referring to the latter, several theories have been proposed, a few of them proved in animal models to support the existence of inflammation in the brains of patients with Huntington's disease. The main assertions of brain inflammation in Huntington's disease are:
- It is the result of a reactive neuronal degeneration process [5].
- It is the result of direct action of mutant huntingtin on microglial physiology, dendritic cells and astrocytes [1],[4],[6].
- It causes the disruption of the blood-brain barrier (BBB) [6] ant thus activates the complement system [1].
In the revised literature, no data about the impact of chronic infections in the pathogenesis of Huntington's disease was found; though there is some evidence claiming its potential to induce neuronal damage through the microglial stadium 3 activation [1]. With this precedent, we intend to raise a fourth hypothetical model about the origin of brain inflammation in patients with Huntington's disease. Our hypothesis suggests that buccal tooth chronic infections -such as periodontitis and its proven capacity [7],[8], to cause several systemic inflammation states- might lead to the transport of inflammatory cytokines to the brain. This could result in a state of persistent low-grade neuroinflammation [7] that may exacerbate the neuronal damage caused by the mutant huntingtin in patients with Huntington's disease [9]. Accordingly, this damage is considered a vital determinant for the onset and progression of neuronal disease. Therefore, buccal tooth chronic infections should be a key factor that both, neurologists and dentists, should take into account in the management of patients with a family history or of those who carry the Huntington's disease-causing mutation.
This review aims to expose the effect of neuroinflammation in the pathogenesis of Huntington's disease, along with raising a hypothetical model to explain the molecular mechanisms by which periodontitis, as a model of chronic dental infection buco, indirectly causes neuroinflammation and neurodegeneration.
Methods
For this review, we appealed to PubMed and SciELO databases, between July 1st and August 1st, 2015. We selected experimental studies, systematic reviews and meta-analyses published between 2000 and 2015 in Spanish and/or English. Search terms used were Huntington's disease, periodontitis, neuroinflammation, neurodegeneration and systemic inflammation.
Results
Neuroinflammation in Huntington's disease
Regarding the first objective, the revised information enables us to note that in Huntington's disease is characteristic the exacerbation in the immune response and the presence of various immune elements due to the accumulation of mutant huntingtin in glial cells and macrophages. The latter are responsible for the autonomic activation of the immune response in the brain [10]. For educational purposes, we will proceed to explain in detail one by one, but it is necessary to understand their actions as a whole and not as separate processes.
Glial cells: microglia and astrocytes
A significant accumulation of activated microglia and astrocytes can be seen in those patients with Huntington's disease and the defective gene even 15 years before the estimated age of onset of the disease [10]. The work of Crotti et al. about the microglia, relates the accumulation of mutant huntingtin with an increased expression and a higher transcription of the determinants of the myeloid lineage, PU.1 and C / EBP. These are responsible, besides the development and function of macrophages and microglia, for determining their regulatory activity through the selection of enhancers and promoters dependent on different transcription factors such as nuclear transcription factor β [4], which is responsible for the increase cytokines and chemokines. Figure 1 illustrates how the expression of mutant huntingtin in microglia produces polarization to the proinflammatory phenotype M1 in this cell, with the consequent release of cytokines and reactive oxygen species, which cause neuronal damage and astrogliosis. Injured neurons release damage associated molecular patterns, thereby enhancing the activation of microglial cells and thus neuroinflammation and its effects on glial cells [1].
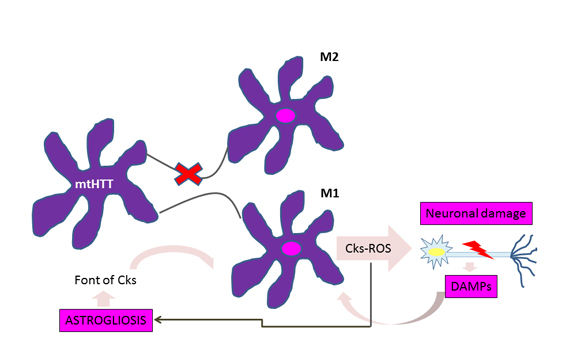
Figure 1: Vicious circle of microglial activation in Huntington's disease.
For astrocytes, murine models show that the accumulation of mutant huntingtin reduces membrane expression of a potassium channel (Kir4.1) generating depolarization and increased levels of extracellular K+ ion, which affects the removal of glutamate within the extracellular space causing excitotoxicity in neuronal tissue [11],[12] (Figure 2). This decrease in the neuroprotection provided by astrocytes, correlates with the functional role of neuroinflammation in Huntington's disease [1]. To this is added the astrogliosis (hypertrophy and increased proliferation of astrocytes) suffered by these cells as a result of proinflammatory mediators secreted by reactive microglia, and the aberrant activation of nuclear transcription factor β, exacerbating the inflammatory response and favoring neurodegeneration, [3] secreting more proinflammatory cytokines that activate more microglial cells, creating a progressive vicious circle [13] (Figure 1). Figure 2 illustrates how the accumulation of mutant huntingtin in astrocytes generates a down-regulation of a potassium channel (Kir4.1), with the consequent membrane depolarization therein. This increases K+ extracellular levels, affecting glutamate removal from the extracellular space and generating excitotoxicity in neuronal tissue [12].
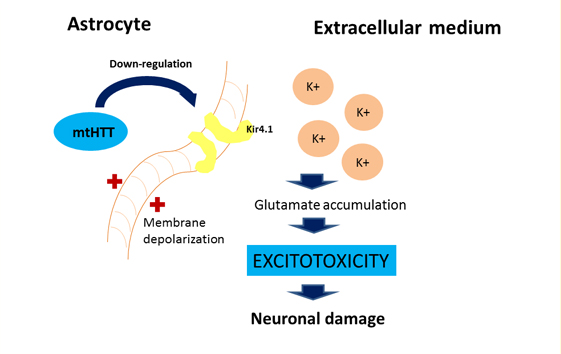
Figure 2: Effect of mutant huntingtin on the astrocyte's physiology
Macrophages
As well as microglia, macrophages phenotype may be type M1, proinflammatory, or M2 type, anti-inflammatory, changing to one or the other depending on the stage of the disease [14].
Macrophages are responsible for cytokine production peripherally, which cross the blood-brain barrier and activate microglia. Its activity is altered in various neurodegenerative diseases such as Huntington's disease, being that an impaired macrophage response to chemoattractants and, consequently, a reduced migration to inflammation sites in patients with this condition has been described. This would be associated with an increased secretion of proinflammatory cytokines. It is presumed that this is because of the accumulation of mutant huntingtin, which would use the same mechanisms as in microglia to interfere with the normal functioning of these cells [1]. There would also be a polarization towards the M1 phenotype, producing a significant reduction in the levels of the transforming growth factor β1, which is produced by the M2 phenotype and has neuroprotective properties, participating in the regulation of microglial activation. Thus, plasma levels are also predictive to determine the age of onset of the disease [14].
Complement system
Being one of the most important elements of the innate immune system and a connection point between it and the adaptive immune system, the role of the complement system is of particular interest in Huntington's disease. In the central nervous system, most of the same components and receptors are expressed in both glia cells and neurons and can be activated by various proteins, including mutant huntingtin. This triggers a cascade of processes such as the secretion of proinflammatory cytokines, attraction of macrophages, increased antigens phagocytosis and cell lysis, among others [1].
Cytokines and chemokines
As already mentioned levels of proinflammatory cytokines and chemokines are increased in Huntington's disease and are responsible for the activation of microglia and astrogliosis, as well as for leukocyte migration from the periphery and the dysfunctional blood-brain barrier [14]. Of particular interest are the IL-6, ISLA-1β, TNF-α [1] and five chemokines (eotaxin-3, MIP-1β, eotaxin, MCP-1 and MCP-4) [15]. It should be noted that the IL-1β secreted by several cell types such as dendritic cells, directly induces neurotoxicity by the activation of tyrosine kinase dependent signaling pathways and the phosphorylation of N-methyl-D-aspartate receptors involved in nuclear transcription factor β pathway [1]. High levels of IL-1β and TNF-α were found in neurodegenerative diseases, altering neuronal function and affecting the concentration of neurotransmitters such as glutamate [16], whose excess, as already mentioned, leads to neurotoxicity [11],[12]. It has also been observed in vitro that the combination of IL-1β and TNF-α produces neuronal death and apoptosis [16].
Role of the adaptive immune system
While the immune response involves a balanced interaction between the innate immune system and the adaptive immune system, according to Ellrichmann et al., it has not been possible to establish the role of the adaptive immune system in Huntington's disease yet. It is suggested that the dendritic cells, T cells and certain specific cytokines of this system; could be involved since the accumulation of mutant huntingtin in dendritic cells cause its constant activation. These in turn, activate T cells, which segregate more cytokines, exacerbating the inflammatory process [1].
Inflammation and blood-brain barrier
Chronic inflammation at peripheral and / or interstitial level would affect the blood-brain barrier capillary wall, allowing the entry to the central nervous system of amyloid peptide [7], inflammatory cells and serum proteins such as chemokines and cytokines [17], aggravating and complementing all the above.
Neuroinflammation and amyloid proteins formation
It has been reported that neuroinflammation induces hyperphosphorylation of tau protein [7] with the consequent formation of oligomeric intermediates and neurofibrillary tangles. Which, in patients with Huntington's disease, interact with fragments of mutant huntingtin forming protein aggregates that produce neuronal dysfunction and neurodegeneration [9]. Additionally, brain inflammation can increase amyloidogenesis raising proteolysis of amyloid peptide precursor, resulting in an increase in the concentration of amyloid monomers or dimers. These ones, with time, would form oligomeric intermediaries, which have proved to be neurotoxic in animal models, leading to neuronal degeneration through the activation of apoptotic death pathways [18].
Chronic periodontitis: a systemic inflammatory disease of low grade
Regarding the second objective set, the revised information defined periodontitis as a destructive, polymicrobial and inflammatory disease that compromises the protective tissues and dental insertion (gingival connective tissue attached to the root surface, periodontal ligament, cementum and adjacent alveolar bone) [8]. It is clinically characterized by bleeding and purulent discharge from the gums, progressive deepening of the gingival sulcus (referred to as periodontal pocket), oral halitosis, dental diastema and dental mobility in advanced stages [7]. It is considered the leading cause of tooth loss [8].
Dental plaque is the primary source of periodontitis, consisting predominantly of Gram-negative and anaerobic bacteria. However, the microbial factor is not sufficient to mediate the progressive destruction of periodontal tissue, being essential to that end, the host response, which ultimately will determine the intensity of tissue destruction. Genetical and environmental or acquired risk factors shall determine the way in which the host responds to the attack by the periodontal-pathogens [7].
The ulcerated lining of the periodontal pocket is a gateway to harmful bacteria and their products into the systemic circulation. It is reported that the total surface area of the ulcerated coating periodontal pocket in patients with severe periodontitis is approximately 15 to 20 square centimeters [7], which allows the entry of bacteria and harmful products to the systemic circulation through three mechanisms [19]:
- Metastatic infection or bacteremia: microorganisms that enter the bloodstream are not eliminated and spread.
- Metastatic damage: lipopolysaccharides and endotoxins which are lethal to the cells are released.
- Metastatic inflammation: by antigen antibody reactions and the release of chemical mediators.
This alters the character of the periodontitis from a local disease to a systemic disorder, capable of supporting a "low-grade systemic inflammation" [7].
Link between periodontitis and Huntington's disease
Periodontitis as a systemic low-grade disease can be a determining factor in the initiation and progression of Huntington's disease, through two potential mechanisms (Figure 3):
- Systemic inflammation preceded by periodontitis.
- Direct viral and bacterial influence.
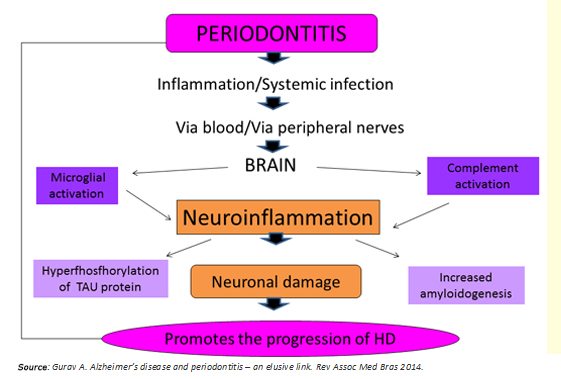
Figure 3: Link between periodontitis and Huntington's disease.
Regarding the first mechanism, bacteria and the host response, raise serum levels of proinflammatory cytokines [7],[8], establishing a state of systemic inflammation that compromises the integrity of the blood-brain barrier. Previously, this would be altered per se in Huntington's disease [6], facilitating the access of these molecules to the brain. They would activate microglia cells, leading to maturation stage 3 [1],[10], thus adopting a proinflammatory phenotype M1 to release cytokines such as IL-1B, TNF-a, IL-6, and other inflamogenic molecules that activate the complement system in the brain [8]. This process generates a state of "neuro persistent low-grade inflammation", capable of causing neuronal death in two ways: activation of apoptotic pathways [16] and increasing the concentration of the neurotransmitter glutamate producing neurotoxicity [11],[12].
The second mechanism involves the brain invasion by residential bacteria and viruses of the dental plaque, which would be transported, or through blood or via peripheral nerves [7]. The research supporting this second proposed mechanism are:
- Riviere et al. isolated spirochetes species such as Treponema denticola, Treponema pectinovorum, Treponema vincentii, Treponema amylovorum, Treponema maltophilum, Treponema socranskii and Treponema medium, in the brain of patients with Alzheimer's disease, using a specific PCR (polymerase chain reaction).It has been speculated that the oral cavity treponemal species may have access to the cerebral cortex via the trigeminal nerve [20]. In this regard, one study found a statistically significant association between spirochetes and Alzheimer's disease, after detecting treponemes in 93.7% of the patients with Alzheimer's disease compared to 33.3% of the control subjects [21].
- Poole et al. detected Porphyromonas gingivalis components in the brains of patients with Alzheimer's disease [22]. Thanks to this discovery, the association of periodontal disease with age of onset and perpetuation of inflammation in Alzheimer's disease has been proposed. It has been proved that the Pophyromonas gingivalis is an expert evading the host immune response, using for it several mechanisms including:
a) Natural ability to form biofilm [23].
b) Degrading complement components through the production of proteases [24]..
c) Recruiting host regulatory proteins such as factor H and C4 protein [25].
d) Ability to adhere to erythrocytes via complement receptor 1 (CR1), which allows the bacteria to pass undetected
by circulating phagocytes, and be transported via systemic circulation accessing remote body organs in individuals
with susceptibility for developing inflammatory diseases [26].
- Rao Singh et al. considered Porphyromonas gingivalis to be a key inflamophilic pathogen, able to establish communities with other microbes, exacerbating inflammation in the host. They also suggest that periodontal pathogens entering the brain produce inflammation through microglial and complement system activation [8].
In synthesis, chronic periodontitis would be a potential source of neuronal damage through its proven ability to generate low-grade systemic inflammation and spread to the brain periodontal pathogens through hematologic and neural routes. Both situations predominantly activate the innate immune system by microglial M1 phenotype polarization and alternative pathway activation of the complement system, which ultimately enhances microglial activation with consequent release of cytokines and reactive oxygen species (Figure 4). According to Ellrichmann et al., this would produce astrogliosis (hypertrophy and an increase in the astrocyte proliferation in response to proinflammatory stimuli) [3]. Thereby, both systemic inflammation and the same Huntington's disease exacerbate the promoted brain inflammation. This neuroinflammation produces neuronal injury due to:
- Activation of apoptotic pathways.
- TAU protein hyperphosphorylation.
- Increased amyloidogenesis.
Injured neurons would release damage associated molecular patterns at later stages of the death process, promoting microglial activation in the brain, and therefore neuronal injury, leading to cognitive impairment [27] (Figures 1 and 3).
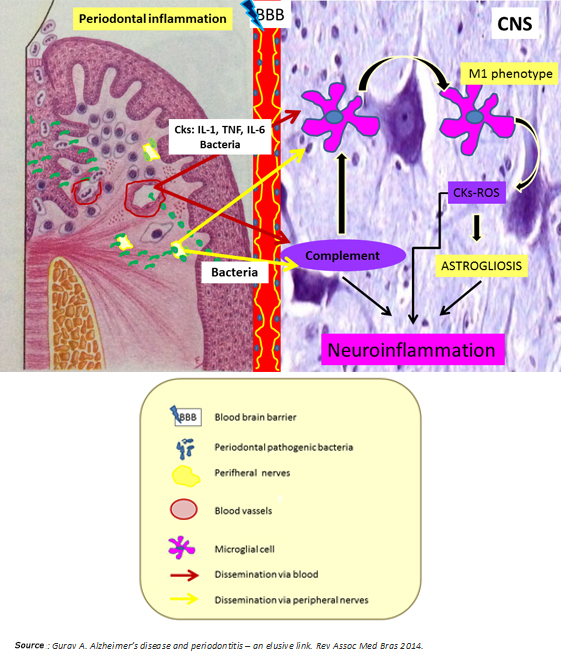
Figure 4: Nexus between periodontitis and Huntington's disease.
Final thoughts
Thanks to the development of molecular biology, it could be possible to identify the cause of Huntington's disease. However, the exact mechanism that causes neuronal damage and cognitive impairment in these patients is still unknown. Recent reports give the neuroinflammation a key role in the pathogenesis of Huntington's disease [4],[6]. All the knowledge currently available on this subject results from animal models, which attribute the mutant huntingtin the responsibility to promote and orchestrate the inflammatory stage in the brains of these patients through innate and adaptive immune mechanisms [1]. Moreover, this neuroinflammation orchestrated by mutant huntingtin may be exacerbated by systemic infections capable of generate and maintain a peripheral low-grade inflammatory state.
We propose that chronic oral and dental infections as periodontal disease could be a determining factor in the onset and progression of Huntington's disease, due to the reviewed mechanisms. However, we remark that there are no animal models that confirm this hypothesis, so this issue should be considered for future research on this disease. If this association were demonstrated, it would be possible to make a multidisciplinary approach to the patients with Huntington's disease, to the patients with a family history or to those who carry the causing mutation of the disease. Because an early diagnosis of infectious oral pathologies with systemic impact in this group of patients would control the degree of cognitive impairment. So an early debut and the fatal clinical course of this neuronal disease could be prevented.
Conclusions
Various mechanisms are responsible for generating inflammation in the brains of patients with Huntington's disease, acquiring special prominence the innate immune mechanisms. Chronic oral and dental infections as periodontitis may be an exacerbating factor of inflammation which itself accompanies this disease.
Before this background, we suggest to investigate though preclinical and clinical models the association between periodontitis and Huntington's disease in order to delay the onset of this disease implementing additional care and prevention measures from the dentist's place.
The same model we propose could be expanded to other common chronic inflammatory conditions. Once proven, it would allow acting on the quality of life improvement of the patients.
Notes
We, the authors, take responsibility for the content and originality of the illustrations in this work, and we claim to have no conflict of interests.
From the editor
The authors originally submitted this article in Spanish and afterwards translated it into English. The Journal has not copyedited this version.
Conflicts of interests
The authors have completed the conflict of interests declaration form from the ICMJE, and declared not having any conflict of interests with the matter dealt herein. Forms can be requested to the responsible author or the editorial direction of the Journal.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.

La enfermedad de Hungtinton es un trastorno neurodegenerativo, causado por la expansión de un triplete de citosina, adenina, guanina en el gen de la huntingtina. Se manifiesta con un deterioro físico, cognitivo y psiquiátrico a distintas edades en el adulto, con un pronóstico fatal. Además del número de repeticiones del triplete, existirían otros factores que explicarían el inicio de esta enfermedad a más temprana edad. Se sabe que la neuroinflamación es un protagonista en los trastornos neurodegenerativos, no siendo la enfermedad de Huntington una excepción. La neuroinflamación exacerba el dańo neuronal producido por la mutación, al existir activación aberrante de la célula microglía, disfunción de astrocitos y células dendríticas; compromiso de la barrera hematoencefálica y activación de complemento, todas ellas como efecto directo e indirecto de la mutante y otros estímulos como infecciones crónicas. Es el interés del presente trabajo analizar la periodontitis, como modelo de infección bucodental crónica y fuente de inflamación sistémica. Hipotetizamos que el potencial rol de la periodontitis en la enfermedad de Huntington y los mecanismos por los cuales contribuiría a la manifestación temprana y progreso de dicha enfermedad, para lo cual se consideraron revisiones sistemáticas, metanálisis y estudios experimentales publicados tanto en espańol como en inglés obtenidos del PubMed y SciELO. Son diversos los mecanismos que generan inflamación en el cerebro de estos pacientes, adquiriendo especial protagonismo los mecanismos de la inmunidad innata. Las infecciones buco dentarias crónicas, como la enfermedad periodontal, pueden constituir un factor exacerbante de la neuroinflamación que per se asocia la enfermedad de Huntington.
Authors:
María Lourdes Rodríguez Coyago[1,2,3], Victoria Emilia Sánchez Temińo[1,4]
Affiliation:
[1] Facultad de Bioquímica y Farmacia, Universidad de Buenos Aires, Buenos Aires, Argentina
[2] Centro de Micología IMPAM, Facultad de Medicina, Universidad de Buenos Aires, Buenos Aires, Argentina
[3] Facultad de Odontología, Universidad de Cuenca, Cuenca, Ecuador
[4] Centro de Investigación en Porfirias y Porfirinas (CIPYP), Hospital de Clínicas, Buenos Aires, Argentina
E-mail: malourdes84@hotmail.com
Author address:
[1] Avenida Callao 650
Capital Federal
Buenos Aires
Argentina
Citation: Rodríguez Coyago ML, Sánchez Temińo VE. Periodontitis determining the onset and progression of Huntington's disease: review of the literature. Medwave 2015 Oct;15(9):e6293 doi: 10.5867/medwave.2015.09.6293
Submission date: 23/8/2015
Acceptance date: 19/10/2015
Publication date: 27/10/2015
Origin: not requested
Type of review: reviewed by three external peer reviewers, double-blind
Comments (0)
We are pleased to have your comment on one of our articles. Your comment will be published as soon as it is posted. However, Medwave reserves the right to remove it later if the editors consider your comment to be: offensive in some sense, irrelevant, trivial, contains grammatical mistakes, contains political harangues, appears to be advertising, contains data from a particular person or suggests the need for changes in practice in terms of diagnostic, preventive or therapeutic interventions, if that evidence has not previously been published in a peer-reviewed journal.
No comments on this article.
To comment please log in
Medwave provides HTML and PDF download counts as well as other harvested interaction metrics. There may be a 48-hour delay for most recent metrics to be posted.
- Ellrichmann G, Reick C, Saft C, Linker R. The Role of the Immune System in Huntington’s Disease. Clin Dev Immunol. 2013;1(1):541259. | CrossRef |
- Bates GP. History of genetic disease: the molecular genetics of Huntington disease - a history. Nat Rev Genet. 2005 Oct;6(10):766-73. | PubMed |
- Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y. A critical role of astrocyte-mediated nuclear factor-κB- dependent inflammation in Huntington's disease. Hum Mol Genet. 2013 May 1;22(9):1826-42. | CrossRef | PubMed |
- Crotti A, Benner C, Kerman BE, Gosselin D, Lagier-Tourenne C, Zuccato C, et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014 Apr;17(4):513-21. | CrossRef | PubMed |
- Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014 Jul;14(7):463-77. | CrossRef | PubMed |
- Hsiao HY, Chen YC, Huang CH, Chen CC, Hsu YH, Chen HM, et al. Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann Neurol. 2015 Aug;78(2):178-92. | CrossRef | PubMed |
- Gurav AN. Alzheimer's disease and periodontitis--an elusive link. Rev Assoc Med Bras. 2014 Mar-Apr;60(2):173-80. | PubMed |
- Singhrao SK, Harding A, Poole S, Kesavalu L, Crean S. Porphyromonas gingivalis Periodontal Infection and Its Putative Links with Alzheimer's Disease. Mediators Inflamm. 2015;2015:137357. | CrossRef | PubMed |
- Vuono R, Winder-Rhodes S, de Silva R, Cisbani G, Drouin-Ouellet J, Spillantini MG, et al. The role of tau in the pathological process and clinical expression of Huntington's disease. Brain. 2015 Jul;138(Pt 7):1907-18. | CrossRef | PubMed |
- Crotti A, Glass CK. The choreography of neuroinflammation in Huntington's disease. Trends Immunol. 2015 Jun;36(6):364-73. | CrossRef | PubMed |
- Chan CS, Surmeier DJ. Astrocytes go awry in Huntington's disease. Nat Neurosci. 2014 May;17(5):641-2. | PubMed |
- Khakh BS, Sofroniew MV. Astrocytes and Huntington's disease. ACS Chem Neurosci. 2014 Jul 16;5(7):494-6. | CrossRef | PubMed |
- Jansen AH, Reits EA, Hol EM. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front Mol Neurosci. 2014 Aug 8;7:73. | CrossRef | PubMed |
- Di Pardo A, Alberti S, Maglione V, Amico E, Cortes EP, Elifani F, et al. Changes of peripheral TGF-β1 depend on monocytes-derived macrophages in Huntington disease. Mol Brain. 2013 Dec 13;6:55. | CrossRef | PubMed |
- Wild E, Magnusson A, Lahiri N, Krus U, Orth M, Tabrizi S, et al. Abnormal peripheral chemokine profile in Huntington’s disease. PLoS curr. 2011;1(1). | CrossRef |
- Ye L, Huang Y, Zhao L, Li Y, Sun L, Zhou Y, et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem. 2013 Jun;125(6):897-908. | CrossRef | PubMed |
- Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012 Jul;122(7):2454-68. | CrossRef | PubMed |
- Guglielmotto M, Monteleone D, Piras A, Valsecchi V, Tropiano M, Ariano S, et al. Aβ1-42 monomers or oligomers have different effects on autophagy and apoptosis. Autophagy. 2014 Oct 1;10(10):1827-43. | CrossRef | PubMed |
- Peńa M, Peńa L, Díaz A, Torres D, Lao Salas N. La enfermedad periodontal como riesgo de enfermedades sistémicas. Rev Cubana Estomatol. 2008;45(1). | Link |
- Miklossy J. Alzheimer's disease - a neurospirochetosis. Analysis of the evidence following Koch's and Hill's criteria. J Neuroinflammation. 2011 Aug 4;8:90. | CrossRef | PubMed |
- Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer's disease brain tissue. J Alzheimers Dis. 2013;36(4):665-77. | CrossRef | PubMed |
- Darveau RP, Hajishengallis G, Curtis MA. Porphyromonas gingivalis as a potential community activist for disease. J Dent Res. 2012 Sep;91(9):816-20. | CrossRef | PubMed |
- Mahtout H, Chandad F, Rojo JM, Grenier D. Porphyromonas gingivalis mediates the shedding and proteolysis of complement regulatory protein CD46 expressed by oral epithelial cells. Oral Microbiol Immunol. 2009 Oct;24(5):396-400. | CrossRef | PubMed |
- Potempa M, Potempa J, Okroj M, Popadiak K, Eick S, Nguyen KA, et al. Binding of complement inhibitor C4b-binding protein contributes to serum resistance of Porphyromonas gingivalis. J Immunol. 2008 Oct 15;181(8):5537-44. | PubMed |
- Belstrřm D, Holmstrup P, Damgaard C, Borch TS, Skjřdt MO, Bendtzen K, et al. The atherogenic bacterium Porphyromonas gingivalis evades circulating phagocytes by adhering to erythrocytes. Infect Immun. 2011 Apr;79(4):1559-65. | CrossRef | PubMed |
- Cherry JD, Olschowka JA, O'Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014 Jun 3;11:98. | CrossRef | PubMed |
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000 May-Jun;21(3):383-421. | PubMed |
Ellrichmann G, Reick C, Saft C, Linker R. The Role of the Immune System in Huntington’s Disease. Clin Dev Immunol. 2013;1(1):541259. | CrossRef |Bates GP. History of genetic disease: the molecular genetics of Huntington disease - a history. Nat Rev Genet. 2005 Oct;6(10):766-73. | PubMed |Hsiao HY, Chen YC, Chen HM, Tu PH, Chern Y. A critical role of astrocyte-mediated nuclear factor-κB- dependent inflammation in Huntington's disease. Hum Mol Genet. 2013 May 1;22(9):1826-42.
| CrossRef | PubMed |Crotti A, Benner C, Kerman BE, Gosselin D, Lagier-Tourenne C, Zuccato C, et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014 Apr;17(4):513-21.
| CrossRef | PubMed |Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014 Jul;14(7):463-77. | CrossRef | PubMed |Hsiao HY, Chen YC, Huang CH, Chen CC, Hsu YH, Chen HM, et al. Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann Neurol. 2015 Aug;78(2):178-92.
| CrossRef | PubMed |Gurav AN. Alzheimer's disease and periodontitis--an elusive link. Rev Assoc Med Bras. 2014 Mar-Apr;60(2):173-80. | PubMed |Singhrao SK, Harding A, Poole S, Kesavalu L, Crean S. Porphyromonas gingivalis Periodontal Infection and Its Putative Links with Alzheimer's Disease. Mediators Inflamm. 2015;2015:137357.
| CrossRef | PubMed |Vuono R, Winder-Rhodes S, de Silva R, Cisbani G, Drouin-Ouellet J, Spillantini MG, et al. The role of tau in the pathological process and clinical expression of Huntington's disease. Brain. 2015 Jul;138(Pt 7):1907-18. | CrossRef | PubMed |Crotti A, Glass CK. The choreography of neuroinflammation in Huntington's disease. Trends Immunol. 2015 Jun;36(6):364-73. | CrossRef | PubMed |Chan CS, Surmeier DJ. Astrocytes go awry in Huntington's disease. Nat Neurosci. 2014 May;17(5):641-2. | PubMed |Khakh BS, Sofroniew MV. Astrocytes and Huntington's disease. ACS Chem Neurosci. 2014 Jul 16;5(7):494-6. | CrossRef | PubMed |Jansen AH, Reits EA, Hol EM. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front Mol Neurosci. 2014 Aug 8;7:73. | CrossRef | PubMed |Di Pardo A, Alberti S, Maglione V, Amico E, Cortes EP, Elifani F, et al. Changes of peripheral TGF-β1 depend on monocytes-derived macrophages in Huntington disease. Mol Brain. 2013 Dec 13;6:55. | CrossRef | PubMed |Wild E, Magnusson A, Lahiri N, Krus U, Orth M, Tabrizi S, et al. Abnormal peripheral chemokine profile in Huntington’s disease. PLoS curr. 2011;1(1). | CrossRef |Ye L, Huang Y, Zhao L, Li Y, Sun L, Zhou Y, et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem. 2013 Jun;125(6):897-908. | CrossRef | PubMed |Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012 Jul;122(7):2454-68. | CrossRef | PubMed |Guglielmotto M, Monteleone D, Piras A, Valsecchi V, Tropiano M, Ariano S, et al. Aβ1-42 monomers or oligomers have different effects on autophagy and apoptosis. Autophagy. 2014 Oct 1;10(10):1827-43. | CrossRef | PubMed |Peńa M, Peńa L, Díaz A, Torres D, Lao Salas N. La enfermedad periodontal como riesgo de enfermedades sistémicas. Rev Cubana Estomatol. 2008;45(1). | Link |Miklossy J. Alzheimer's disease - a neurospirochetosis. Analysis of the evidence following Koch's and Hill's criteria. J Neuroinflammation. 2011 Aug 4;8:90. | CrossRef | PubMed |Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer's disease brain tissue. J Alzheimers Dis. 2013;36(4):665-77. | CrossRef | PubMed |Darveau RP, Hajishengallis G, Curtis MA. Porphyromonas gingivalis as a potential community activist for disease. J Dent Res. 2012 Sep;91(9):816-20. | CrossRef | PubMed |Mahtout H, Chandad F, Rojo JM, Grenier D. Porphyromonas gingivalis mediates the shedding and proteolysis of complement regulatory protein CD46 expressed by oral epithelial cells. Oral Microbiol Immunol. 2009 Oct;24(5):396-400. | CrossRef | PubMed |Potempa M, Potempa J, Okroj M, Popadiak K, Eick S, Nguyen KA, et al. Binding of complement inhibitor C4b-binding protein contributes to serum resistance of Porphyromonas gingivalis. J Immunol. 2008 Oct 15;181(8):5537-44.
| PubMed |Belstrřm D, Holmstrup P, Damgaard C, Borch TS, Skjřdt MO, Bendtzen K, et al. The atherogenic bacterium Porphyromonas gingivalis evades circulating phagocytes by adhering to erythrocytes. Infect Immun. 2011 Apr;79(4):1559-65. | CrossRef | PubMed |Cherry JD, Olschowka JA, O'Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014 Jun 3;11:98.
| CrossRef | PubMed |Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000 May-Jun;21(3):383-421. | PubMed |





 Research papers
Research papersSystematization of initiatives in sexual and reproductive health about good practices criteria in response to the COVID-19 pandemic in primary health care in Chile
Clinical, psychological, social, and family characterization of suicidal behavior in Chilean adolescents: a multiple correspondence analysis
