
Key Words: pulmonary alveolar proteinosis, bronchoalveolar lavage, interstitial lung disease
Abstract
INTRODUCTION
Pulmonary alveolar proteinosis is a rare, diffuse interstitial lung disease, characterized by alveolar obstruction due to the accumulation of pulmonary surfactant.
CLINICAL PRESENTATION
A 30-year-old male with progressively worsening dyspnea and non-productive cough for one year. He was a sugar cane plantation worker and had prior recurrent respiratory infections. Physical exam revealed cyanosis, and bilateral coarse and fine rales. Chest computed tomography showed diffuse crazy paving pattern. Bronchoscopy with bronchoalveolar lavage yielded a foamy, thick whitish material. Cytology revealed lymphocytes and acellular proteinaceous eosinophilic material. Transbronchial biopsy confirmed the diagnosis of pulmonary alveolar proteinosis. Patient met criteria for whole lung lavage, responding favorably to this therapy.
CONCLUSION
Pulmonary alveolar proteinosis is a rare lung disease and important to consider due to the diagnostic and therapeutic challenge it represents.
Introduction
Pulmonary Alveolar Proteinosis is a rare diffuse interstitial disease characterized by alveolar obstruction due to the collection of lung surfactant in the alveolar space, which results in respiratory failure and alterations in pulmonary gas exchange [1],[2],[3]. It was first described by Rosen, Castleman and Liebow in 1958 [4]. Its annual incidence and prevalence are 0.36-0.49 and 3.7-6.2 cases per million respectively with a male to female ratio ranging from 2.1-2.7:1 [5],[6]. It is more frequent in adults between their second and their fifth decade of life; although, it may affect individuals with different ages [7],[8].
Physiologically, pulmonary surfactant reduces superficial alveolar tension, prevents alveolar collapse, and defends the host from microbial pathogens. The pulmonary surfactant catabolism in alveolar macrophages requires the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF). A disruption at this level allows alveolar accumulation of this lipoprotein material, due to the decrease of its macrophage clearance, which explains the pathophysiology of the disease [1],[8].
The clinical presentation is usually insidious, with an indolent subacute course that delays diagnosis [6]. Symptoms are nonspecific as exertional dyspnea with or without cough. Some patients will present with weight loss, haemoptysis or fever. Physical examination is usually normal, though there might be fine rales in chest auscultation, clubbing and cyanosis [5],[6],[9]. The diagnosis is suspected based on the clinical picture and radiological findings. It is confirmed by bronchoscopy with bronchoalveolar lavage, yielding the characteristic foamy milk-like aspect alveolar fluid with amyloid bodies on cytological examination [1],[6].
The therapeutic algorithm in symptomatic patients includes primarily whole lung lavage. When this is successful, a rapid clinical and radiological improvement is noted. However, recurrences are common and repeating the procedure might be required. Another option is to administer granulocyte-macrophage colony-stimulating factor when there is no response to the first treatment or if the patient could not tolerate the procedure [1],[9],[10]. Long-term prognosis on these patients is uncertain, since some patients might remain stable without intervention. Some survival rates are reported up to 65 - 75% five to ten years after initial diagnosis [9]. We will present a case of pulmonary alveolar proteinosis, a diffuse interstitial disease that is unusual and interesting because of its diagnostic and therapeutic challenges.
Case description
A 30 year-old male patient, living in Chocope (North-Coast of Peru), arrived to the emergency department of The Hospital Regional Docente de Trujillo. He complained about progressively worsening dyspnea and intermittent dry cough for one year. Three months before his admission, he experienced subjective fever. He described worsening dyspnea three days before his admission.
Social history included being a professional soccer player from 19 to 25 years of age. He previously worked on a sugarcane plant for six months, two years before his presentation. His past medical history included recurrent respiratory infections treated with antibiotics, with partial remission; and 10 pounds weight lost over a period of 12 months. There was no prior history or exposure to tuberculosis, smoking or alcoholism. Family history was notable for asthma in his paternal grandfather.
Upon physical examination, admission vital signs were the following: blood pressure 120/70 mmHg, heart rate 102 beats per minute, respiratory frequency 31 per minute, temperature (axillary) 38.5°C. Oxygen saturation was 88% and fraction of inspired oxygen (FiO2) was 40%. He was cyanotic, breathing in semi-sitting position, and with adequate state of hydration and nutrition. On lung exam, there was decreased vibration on palpation in both hemithoraces, as well as diffuse fine rales bilaterally. The rest of the physical examination was unremarkable. Laboratory results showed both normal cell blood count and basic metabolic panel, a high lactate dehydrogenase level (LDH) 1,018 mg/dl. Other studies including antinuclear antibodies (ANA) and double strand anti DNA antibodies (Anti-dsDNA) were both negative.
Arterial blood gases analysis, performed on room air revealed partial pressure of carbon dioxide (PCO2) of 26 mmHg, partial pressure of oxygen (PO2): 60 mmHg, bicarbonate (HCO3-): 17 mmol/l, ratio of partial pressure arterial oxygen and fraction of inspired oxygen (PO2/FiO2): 69 and a high arterial-alveolar gradient (A-aDO2: 531). Direct sputum analysis with Ziehl-Neelsen Stain for acid fast bacilli was negative as well as his ELISA for HIV. Chest radiograph revealed bilateral irregular and heterogeneous infiltrates (Figure 1). Computed tomography of the chest with contrast showed a “crazy paving” pattern (Figure 2).
Figure 1. Chest radiograph on admission.
Figure 2. Inpatient computed tomography (CT), chest with contrast.
During hospitalization he received antibiotic treatment with ceftriaxone (2 g Intravenous (IV) / 24 hours for seven days) followed by clindamycin (600 mg IV/ 8 hours for 10 days) plus trimethoprim/sulfamethoxazole (TMP/SMX) (800/160 mg two tablets orally (PO) / 8 hours for 14 days), and prednisone 40 mg every 24-hours PO for 14 days with mild clinical improvement.
Flexible bronchoscopy was performed showing on inspection acute inflammatory signs on both right upper and middle lobes. The bronchoalveolar lavage yielded a milk-like foamy mucinous material (Figure 3). There were no endobronchial abnormalities. Cytology revealed lymphocyte and an acellular eosinophilic proteinaceous material occupying some alveolar spaces. The anatomopathological study of the transbronchial biopsy showed pulmonary alveolar proteinosis, Periodic Acid Schiff (PAS) and Groccott stains were positive and negative respectively.
Having confirmation of pulmonary alveolar proteinosis, the patient underwent whole lung lavage, initially performed in the operating room and then in the intensive care unit. This procedure was repeated in three occasions. Post-procedure, he received Ceftazidime two grams every eight hours intravenously for 10 days; n-Acetylcysteine 600 mg every 12 hours PO for 15 days; and respiratory physiotherapy for 10 days. Clinical improvement followed treatment. Currently, he is followed up in the outpatient Pulmonary Clinic.
Figure 3. Flexible bronchoscopy.
Discussion
Pulmonary alveolar proteinosis is a rare disease characterized by the accumulation of surfactant in the alveoli; this will lead to respiratory distress, myeloid cell dysfunction, and an increased risk of respiratory infections [11],[12]. The very low prevalence of the disease was reported by Huaringa and Francis [2], who conducted a review including 263 cases throughout the world between the years 1988 and 2014 [2]. There has been only one case reported in Peru by Portugal et al [13], studied, diagnosed and treated after transferring the patient to “Pulmonary Consult of Palm Beaches”, Florida-USA, on February 1999 [13].
There are three main forms of pulmonary alveolar proteinosis: Autoimmune or primary, secondary, and congenital. On all these, the decrease of the macrophage clearance of the surfactant is common, leading to its retention on the terminal airways and alveolar space [5],[9],[10]. Physiopathologically, pulmonary alveolar proteinosis may be caused by the granulocyte-macrophage colony-stimulating factor (GM-CSF) signalization interruption due to a high level of anti GM-CSF autoantibodies, representing 90% of the cases. Other causes include, either, genetic mutations on the GM-CSF alpha and beta receptors (hereditary form); or by an underlying disease affecting the function and number of alveolar macrophages (secondary form). The latter may occur as consequence of underlying clinical disorders such as malignant blood neoplasms, dust inhalation, toxics, fumes, gases, or immunosuppression due to infections or drugs [1],[5],[8].
In our case, we suspected a secondary form based on his prior exposure to dust and fumes as a plantation sugarcane worker. Although this was not a determinant factor, because of the lack of studies determining the necessary time exposure to a certain contaminant causing the disease. On the other hand, we cannot exclude an autoimmune form, the most frequent disease presentation [8].
The clinical course of pulmonary alveolar proteinosis is variable, ranging from asymptomatic patients to cases of respiratory failure. Clinical findings are not specific; being cough and dyspnea the most frequent symptoms [7]. However, patients may present with fever and weight loss, hemoptysis is exceptional. On physical exam, the most frequent signs are cyanosis, clubbing and bilateral fine rales [8]. A study performed in China at The First Affiliated Hospital - Wenzhou University of Medicine by Mo et al [5], eleven pulmonary alveolar proteinosis cases were reviewed (seven men and four women). The most common clinical symptoms were gradual onset, progressive dyspnea (91%), cough (73%), fever (4%) and chest pain (18%). Nine percent of the patients were asymptomatic. On physical exam, fine rales were found in 55%, clubbing in 45%, cyanosis in 18% of the patients; and 18% were found normal on physical examination [5]. This case presented as symptoms: cough, dyspnea and weight loss; and as signs: cyanosis, fever and bilateral cracklings.
Among the imaging tests used for the diagnosis we found chest radiographs, showing bilateral perihilar nodular infiltrates, with areas of consolidation. Sometimes they coalesce predominantly on the lung bases, poorly defined mosaic-like and ground glass with “butterfly wings” pattern, mimicking atypical pneumonia or massive pulmonary edema. On high resolution chest computed tomography, diffuse ground glass opacities with septal, intra and interlobar superposition of the thickening; a pattern known as “crazy paving” is observed, which is characteristic of this disease but not pathognomonic [7],[10],[11]. Both chest radiograph and computed tomography scan findings were found in this case.
To confirm diagnosis, it is necessary to perform a transbronchial fiberoptic bronchoscopy guided biopsy, were the pulmonary parenchymal structure is observed. The most notable findings are: alveolar space filling with an acellular, granular, amorphous eosinophilic material which will normally dye positively Schiff’s periodic acid, and Type 2 pneumocyte hyperplasia on the alveolar septa. Moreover, when a bronchoalveolar lavage is performed, milk-like foamy opaque fluid is pathognomonic [1],[9],[14],[15], as it was found on the present reported case.
Differential diagnosis includes those disorders with similar radiological findings. These may be: Pneumocystis jirovecii and Mycoplasma sp pulmonary infections, non-cardiogenic and cardiogenic pulmonary edema, lipoid pneumonia, drug-related hypersensitivity reaction, organizing pneumonia, acute interstitial pneumonia, diffuse alveolar damage overlapped with usual interstitial pneumonitis, alveolar hemorrhagic syndromes, and what used to be called mucinous bronchioloalveolar carcinoma [16].
The most accepted standard treatment is whole lung lavage, with the goal of extracting the proteinaceous material occupying the airway. This procedure is diagnostic and therapeutic. The technique is indicated when there is a definitive histological diagnosis, a PaO2 less than 65 mmHg, an alveolar arterial gradient more than 40 mmHg, a more than 12% shunt and dyspnea during rest [7],[11]. Our patient received the aforementioned treatment, fulfilling the already known criteria, and showing immediate improvement. There are alternative treatments such as subcutaneous or inhaled granulocyte-macrophage colony-stimulating factor (GM-CSF), plasmapheresis and rituximab [8],[10]. These treatments are administered when there is lack of response to whole lung lavage or the patient does not tolerate the conventional treatment. Our patient did not need these treatments.
The prognosis for these patients is uncertain and the precipitating factors are unknown. Different studies have established three disease course patterns: those who remain stable although they will present recurrent symptoms, those who will have spontaneous remission and will respond rapidly days after the conventional treatment, and lastly, those who will have a progressive impairment of the respiratory function. The majority of the cases (72%) will die secondary to respiratory failure or by uncontrolled infectious process (18%), usually during the first year after diagnosis [8]. Our patient had a good clinical response to whole lung lavage, remaining stable and currently being followed as an outpatient.
Conclusion
The present case of pulmonary alveolar proteinosis underlines the relationship of the disease with work exposure, nonspecific symptoms and signs, and characteristic radiologic and tomographic findings; which we need to take into account when we are caring for patients with diffuse interstitial pulmonary disease. Also, it highlights the usefulness of transbronchial biopsy to obtain histological confirmation, and whole lung lavage, as a therapeutic method in the majority of cases. On the other hand, it is worth noting that there are still many aspects of the disease that are unknown, and further research is needed.
Notes
From the editor
The authors originally submitted this article in Spanish and subsequently translated it into English. The Journal has not copyedited this version.
Ethical aspects
The informed consent requested by Medwave has been signed by the patient, a copy of which was forwarded to the editorial board of the Journal.
Declaration of conflicts of interest
The authors have completed the ICMJE Conflict of Interest declaration form, and declare that they have not received funding for the report; have no financial relationships with organizations that might have an interest in the published article in the last three years; and have no other relationships or activities that could influence the published article. Forms can be requested by contacting the author responsible or the editorial management of the Journal.
Financing
The authors declare that there were no external sources of funding.
Note from the editor
The principal or responsible author states that this manuscript is an honest, precise, and transparent transcription of the study reported; that no important aspect from the study has been omitted; and that discrepancies between the study results and those foreseen (if these were relevant) have been registered and explained.
Acknowledgements
The authors would like to thanks to Drs. José Cárdenas-García, Julio Hilario-Vargas and Tony Chávez-Uceda, for carefully reviewing the manuscript and for their most useful suggestions.

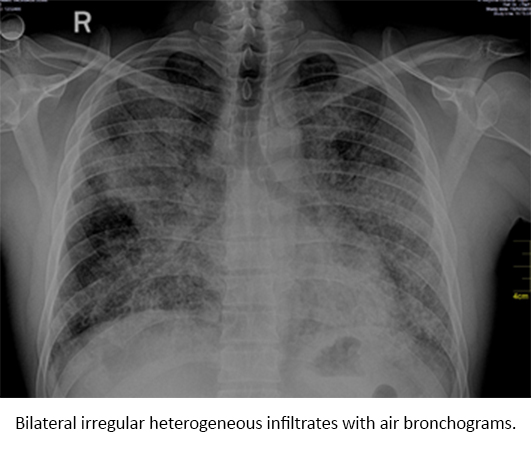 Figure 1. Chest radiograph on admission.
Figure 1. Chest radiograph on admission.
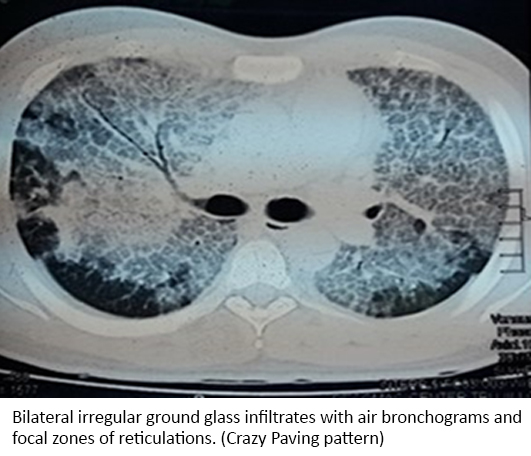 Figure 2. Inpatient computed tomography (CT), chest with contrast.
Figure 2. Inpatient computed tomography (CT), chest with contrast.
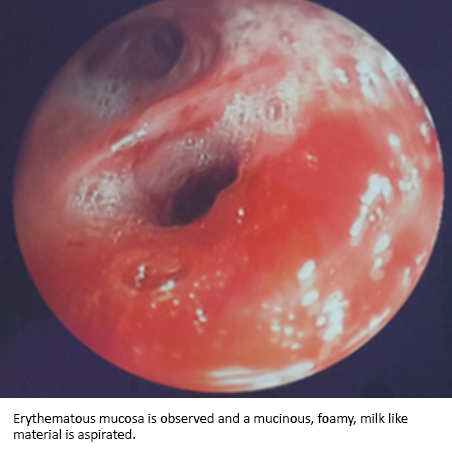 Figure 3. Flexible bronchoscopy.
Figure 3. Flexible bronchoscopy.
 Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
INTRODUCCIėN
La proteinosis alveolar pulmonar es una enfermedad intersticial difusa poco frecuente, en la cual se produce obstrucci¾n alveolar, debido al ac·mulo de surfactante pulmonar.
REPORTE DEL CASO
Var¾n de 30 a±os de edad, present¾ disnea progresiva y tos seca de un a±o de evoluci¾n. Antecedente personal: estibador de ca±a de az·car. Present¾ infecciones respiratorias recurrentes. Al examen fĒsico se encontr¾ cianosis, crepitantes difusos bilaterales y subcrepitantes en bases pulmonares. En la tomografĒa torßcica con contraste se encontr¾ un patr¾n de "empedrado loco". Se realiz¾ videobroncoscopia con lavado broncoalveolar, aspirando material lechoso, espumoso y mucoso. Por citologĒa se encontr¾ linfocitos y material eosinofĒlico proteinßceo acelular. El estudio anatomopatol¾gico de la biopsia transbronquial revel¾ proteinosis alveolar pulmonar. El paciente reuni¾ los criterios para tratamiento con lavado broncoalveolar total. Luego de este procedimiento, evolucion¾ favorablemente.
CONCLUSIėN
La proteinosis alveolar pulmonar constituye una enfermedad importante a considerar, por el desafĒo diagn¾stico y terapķutico que representa.
Authors:
Luis Alberto Concepci¾n-Urteaga[1,2], Luis Alejandro RodrĒguez-Hidalgo[1,2], Jorge Luis Cornejo-Portella[1,2], Oscar Neri Alquizar-Horna[1,2], Daniel Anderson Aguilar-Villanueva[2], Marcio Josķ Concepci¾n-Zavaleta[2], Mario Gustavo Aza±ero-Lujßn[3]
Affiliation:
[1] Centro de Excelencia para el manejo de Tuberculosis ōLuz Caviedes Rojasö, Hospital Regional Docente de Trujillo, Trujillo, Per·
[2] Facultad de Medicina, Universidad Nacional de Trujillo, Trujillo, Per·
[3] Hospital Regional Docente de Trujillo, Trujillo, Per·
E-mail: luisconcepcionurteaga@hotmail.com
Author address:
[1] Avenida Huamßn 242
VĒctor Larco
Trujillo
Per·
Citation: Concepci¾n-Urteaga LA, RodrĒguez-Hidalgo LA, Cornejo-Portella JL, Alquizar-Horna ON, Aguilar-Villanueva DA, Concepci¾n-Zavaleta MJ, et al. Pulmonary alveolar proteinosis: a case report. Medwave 2017 Sep;17(8):e7040 doi: 10.5867/medwave.2017.08.7040
Submission date: 23/6/2017
Acceptance date: 23/8/2017
Publication date: 14/9/2017
Origin: not requested
Type of review: reviewed by four external peer reviewers, double-blind
Comments (0)
We are pleased to have your comment on one of our articles. Your comment will be published as soon as it is posted. However, Medwave reserves the right to remove it later if the editors consider your comment to be: offensive in some sense, irrelevant, trivial, contains grammatical mistakes, contains political harangues, appears to be advertising, contains data from a particular person or suggests the need for changes in practice in terms of diagnostic, preventive or therapeutic interventions, if that evidence has not previously been published in a peer-reviewed journal.
No comments on this article.
To comment please log in
Medwave provides HTML and PDF download counts as well as other harvested interaction metrics. There may be a 48-hour delay for most recent metrics to be posted.
- Suzuki T, Trapnell BC. Pulmonary Alveolar Proteinosis Syndrome. Clin Chest Med. 2016 Sep;37(3):431-40. | CrossRef | PubMed |
- Huaringa AJ, Francis WH. Pulmonary alveolar proteinosis: a case report and world literature review. Respirol Case Rep. 2016 Nov 13;4(6):e00201. | CrossRef | PubMed |
- McDonnell MJ, Reynolds C, Tormey V, Gilmartin JJ, Rutherford RM. Pulmonary alveolar proteinosis: report of two cases in the West of Ireland with review of current literature. Ir J Med Sci. 2014 Mar;183(1):123-7. | CrossRef | PubMed |
- Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958 Jun 5;258(23):1123-42. | PubMed |
- Mo Q, Wang B, Dong N, Bao L, Su X, Li Y, et al. The Clinical Clues of Pulmonary Alveolar Proteinosis: A Report of 11 Cases and Literature Review. Can Respir J. 2016;2016:4021928. | CrossRef | PubMed |
- Khan A, Agarwal R. Pulmonary alveolar proteinosis. Respir Care. 2011 Jul;56(7):1016-28. | CrossRef | PubMed |
- Sßnchez-Valadez TI, Carrillo-Mu±oz A, Valero-G¾mez A, MartĒnez-Pķrez S, Pķrez-Rosales A, N·±ez Pķrez-Redondo C, et al. Proteinosis alveolar: Informe de dos casos y comentario bibliogrßfico. NeumologĒa y cirugĒa de t¾rax. 2015;74(4):271-5. | Link |
- Algarin H, Mendoza DA, Molano D, Claro J. Proteinosis alveolar pulmonar: diagn¾stico y manejo en unidad de cuidado intensivo. Reporte de caso. Acta Colombiana de Cuidado Intensivo. 2017;17(1):76-80. | Link |
- Ernst G, Salvado A, Grynblat P, Tabaj G, Garcia O, Cambursano VH, et al. [Pulmonary alveolar proteinosis: role of GM-CSF Antibodies]. Rev Fac Cien Med Univ Nac Cordoba. 2017;74(1):46-50. | PubMed |
- RodrĒguez J. Tratamiento de la proteinosis alveolar primaria del adulto. Arch Bronconeumol. 2015;51(7):344-9. | Link |
- Fijołek J, Wiatr E, Opoka L, Rudziński P, Nierebińska M, Szołkowska M, et al. Atypical image of pulmonary alveolar proteinosis - a case report. Pneumonol Alergol Pol. 2015;83(6):453-6. | CrossRef | PubMed |
- Ul Rehman S, Hammersley JR. Pulmonary Alveolar Proteinosis: Crazy Paving to Whole Lung Lavage. Mayo Clin Proc. 2016 Nov;91(11):1673-674. | CrossRef | PubMed |
- Portugal J, Torres E, DĒaz F, Minauro C. Proteinosis Alveolar Pulmonar. BoletĒn de la Sociedad Peruana de Medicina Interna. 2000;13(1):47-51. | Link |
- Munir Z, Khosa MZ, Qazi MY. Pulmonary Alveolar Proteinosis: A Rare Cause of Respiratory Failure. J Coll Physicians Surg Pak. 2015 Jul;25(7):541-2. | CrossRef | PubMed |
- Yildirim BB, Akgedik R, Akgedik S, Nazaroglu H. Pulmonary alveolar proteinosis in a marble worker. Int J Occup Med Environ Health. 2016;29(5):871-6. | CrossRef | PubMed |
- Chan ED, King TE. Causes, clinical manifestations, and diagnosis of pulmonary alveolar proteinosis in adults. Walthman, MA: UpToDate; 2017. | Link |
Suzuki T, Trapnell BC. Pulmonary Alveolar Proteinosis Syndrome. Clin Chest Med. 2016 Sep;37(3):431-40. | CrossRef | PubMed |Huaringa AJ, Francis WH. Pulmonary alveolar proteinosis: a case report and world literature review. Respirol Case Rep. 2016 Nov 13;4(6):e00201. | CrossRef | PubMed |McDonnell MJ, Reynolds C, Tormey V, Gilmartin JJ, Rutherford RM. Pulmonary alveolar proteinosis: report of two cases in the West of Ireland with review of current literature. Ir J Med Sci. 2014 Mar;183(1):123-7. | CrossRef | PubMed |Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958 Jun 5;258(23):1123-42. | PubMed |Mo Q, Wang B, Dong N, Bao L, Su X, Li Y, et al. The Clinical Clues of Pulmonary Alveolar Proteinosis: A Report of 11 Cases and Literature Review. Can Respir J. 2016;2016:4021928. | CrossRef | PubMed |Khan A, Agarwal R. Pulmonary alveolar proteinosis. Respir Care. 2011 Jul;56(7):1016-28. | CrossRef | PubMed |Sßnchez-Valadez TI, Carrillo-Mu±oz A, Valero-G¾mez A, MartĒnez-Pķrez S, Pķrez-Rosales A, N·±ez Pķrez-Redondo C, et al. Proteinosis alveolar: Informe de dos casos y comentario bibliogrßfico. NeumologĒa y cirugĒa de t¾rax. 2015;74(4):271-5. | Link |Algarin H, Mendoza DA, Molano D, Claro J. Proteinosis alveolar pulmonar: diagn¾stico y manejo en unidad de cuidado intensivo. Reporte de caso. Acta Colombiana de Cuidado Intensivo. 2017;17(1):76-80. | Link |Ernst G, Salvado A, Grynblat P, Tabaj G, Garcia O, Cambursano VH, et al. [Pulmonary alveolar proteinosis: role of GM-CSF Antibodies]. Rev Fac Cien Med Univ Nac Cordoba. 2017;74(1):46-50. | PubMed |RodrĒguez J. Tratamiento de la proteinosis alveolar primaria del adulto. Arch Bronconeumol. 2015;51(7):344-9. | Link |Fijołek J, Wiatr E, Opoka L, Rudziński P, Nierebińska M, Szołkowska M, et al. Atypical image of pulmonary alveolar proteinosis - a case report. Pneumonol Alergol Pol. 2015;83(6):453-6. | CrossRef | PubMed |Ul Rehman S, Hammersley JR. Pulmonary Alveolar Proteinosis: Crazy Paving to Whole Lung Lavage. Mayo Clin Proc. 2016 Nov;91(11):1673-674. | CrossRef | PubMed |Portugal J, Torres E, DĒaz F, Minauro C. Proteinosis Alveolar Pulmonar. BoletĒn de la Sociedad Peruana de Medicina Interna. 2000;13(1):47-51. | Link |Munir Z, Khosa MZ, Qazi MY. Pulmonary Alveolar Proteinosis: A Rare Cause of Respiratory Failure. J Coll Physicians Surg Pak. 2015 Jul;25(7):541-2. | CrossRef | PubMed |Yildirim BB, Akgedik R, Akgedik S, Nazaroglu H. Pulmonary alveolar proteinosis in a marble worker. Int J Occup Med Environ Health. 2016;29(5):871-6. | CrossRef | PubMed |Chan ED, King TE. Causes, clinical manifestations, and diagnosis of pulmonary alveolar proteinosis in adults. Walthman, MA: UpToDate; 2017. | Link |





 Research papers
Research papersSystematization of initiatives in sexual and reproductive health about good practices criteria in response to the COVID-19 pandemic in primary health care in Chile
Clinical, psychological, social, and family characterization of suicidal behavior in Chilean adolescents: a multiple correspondence analysis
