
Key Words: anophthalmia, congenital heart disease, cor triatriatum, pulmonary hypertension, children
Resumen
La anoftalmía congénita clínica es la ausencia uni o bilateral del globo ocular, que se presenta de forma aislada o como parte de un síndrome. Tiene muy baja prevalencia y su etiología es heterogénea. La presencia conjunta de malformaciones cardíacas congénitas complejas también es poco frecuente. La asociación de anoftalmía congénita con cardiopatías congénitas es más rara aún, la etiología de tales asociaciones no es bien comprendida todavía. Se reporta el caso de una paciente que presentó la muy infrecuente asociación de anoftalmía bilateral, múltiples malformaciones cardíacas e hipertensión arterial pulmonar grave.
Introducción
La anoftalmía congénita clínica es la ausencia uni o bilateral del globo ocular, que se presenta de forma aislada o como parte de un síndrome. La prevalencia de anoftalmía y microftalmía es de 3 y 14 por 100 000 nacimientos respectivamente, no obstante la prevalencia combinada puede llegar hasta 30 por 100 000 nacimientos [1],[2],[3], y su etiología es heterogénea. Aunque la anoftalmía se evidencia clínicamente, el estudio histológico puede presentar residuo ectodérmico. Ello da lugar a denominaciones tales como anoftalmía verdadera, anoftalmía clínica y microftalmía extrema.
Algunas cardiopatías congénitas, tanto derechas como izquierdas, tienen baja prevalencia y que estas se presenten en forma conjunta es muy poco frecuente. La asociación de patologías congénitas tales como anoftalmía y cardiopatías complejas ha sido escasamente comunicada [1],[4],[5],[6],[7], la etiología de tal asociación todavía no se ha dilucidado totalmente.
Este es el informe del caso de una paciente que presentó la rara asociación de anoftalmía clínica bilateral, múltiples malformaciones cardíacas tanto derechas como izquierdas e hipertensión arterial pulmonar.
Presentación del caso
Como es política del comité de ética en investigación de nuestra institución, el siguiente comunicado cuenta con el consentimiento informado firmado por los padres para poder presentar o publicar los datos dentro del contexto científico, sin manifestar la identidad de la paciente.
En la unidad de cuidados intensivos neonatal se recibió una niña de ocho días de edad, peso 3,6 kilogramos (percentil: 50), talla 51 centímetros (percentil: 50) y perímetro cefálico 35 centímetros (percentil: 50), transferida de un hospital rural. El período prenatal fue aparentemente normal, la madre no reportó exposición a infecciones, alcohol, tabaco o drogas durante el embarazo. La paciente nació a término, con escala Apgar 5-7. Los padres son de raza mestiza y primos en primer grado. La madre tiene antecedentes de un aborto espontáneo y una hija fallecida sin causa conocida pocos minutos después de nacer.
Al examen físico la paciente estaba hipoactiva, llanto débil y con dificultad respiratoria, tenía facies dismórfica caracterizada por frente estrecha, hipotelorismo, hendiduras palpebrales pequeñas, anoftalmía bilateral, nariz antevertida y pabellones auriculares de implantación baja e hirsutismo. Los genitales externos femeninos tenían morfología normal y la mano derecha presentó polidactilia preaxial (pulgar bífido). A la auscultación se evidenció soplo sistólico grado 4/6 en borde paraesternal izquierdo superior y murmullo vesicular conservado. Presentó oximetría de pulso 85%, a pesar de recibir oxígeno con casco cerrado (Hood). Debido a que sus signos clínicos y de gasometría fueron compatibles con insuficiencia respiratoria, fue colocada en ventilación mecánica en modo intermitente mandatorio sincronizado.
La radiografía de tórax reveló cardiomegalia moderada e infiltrado pulmonar, que se catalogó como neumonía connatal (Figura 1A). No hubo malformación de la columna vertebral. La ecografía abdominal evidenció poliesplenia, la ecografía renal fue normal. Tanto el tamizaje metabólico neonatal (hipotiroidismo, fenilcetonuria, hiperplasia suprarrenal y galactosemia) como el STORCH (sífilis, toxoplasma, rubeola, citomegalovirus, herpes), fueron negativos. El estudio cromosómico informó cariotipo 46 XX.
La tomografía craneal no informó alteración cerebral y la de órbitas oculares evidenció ausencia de globos oculares con presencia de remanentes quísticos, nervio óptico filiforme, músculos oculares extrínsecos hipoplásicos y criptoftalmos (Figuras 1B y 1C). El electroencefalograma mostró registro del sueño desestructurado, sin actividad epileptiforme.
Figura 1. Exámenes de imagenología.
La ecocardiografía transtorácica informó situs solitus, cor triatriatum sinister (aurícula izquierda divida por una membrana en cámaras proximal y distal) con gradiente moderado transmembrana, comunicación interauricular caudal a la membrana intracameral, hemitronco arterioso y signos de hipertensión pulmonar moderada (Figura 2).
La angiotomografía cardíaca informó cor triatriatum sinister, comunicación interauricular de cuatro milímetros, anomalía de conexión venosa pulmonar mixta (cardíaca y supracardíaca), conducto arterioso permeable grande, la aorta ascendente daba origen a la arteria pulmonar derecha (hemitronco arterioso) y el origen de la arteria coronaria izquierda no fue visualizado (Figura 3).
Figura 3. Angiotomografía cardíaca.
El cateterismo cardíaco corroboró un gran conducto arterioso permeable, cor triatriatum sinister y comunicación interauricular. Además, evidenció ventana aortopulmonar tipo 2 de la clasificación de Mori, anomalía parcial del retorno venoso pulmonar donde las venas pulmonares izquierdas drenaban en la vena innominada a través de la vena vertical y las venas pulmonares derechas en la cámara proximal de la aurícula izquierda. También presentó hipoplasia leve del istmo aórtico, origen de la arteria coronaria izquierda en el tronco arterial pulmonar (ALCAPA, por su sigla en inglés anomalous left coronary artery from the pulmonary artery, o síndrome de Bland-White-Garland) (Figura 4), hipoplasia de las arterias pulmonares con índice de Nakata de 110 mm2 /superficie corporal expresada en metros cuadrados (valor normal 330 +/- 30 mm2/m2) [8] e hipertensión pulmonar grave con test de vasorreactividad negativo.
Figura 4. Cateterismo cardíaco.
A los seis meses de edad se le realizó traqueostomía y gastrostomía, siendo luego transferida a la unidad de cuidados intensivos pediátricos. Durante la hospitalización recibió varios ciclos de antibióticos por neumonías recurrentes, probablemente relacionadas con la ventilación mecánica prolongada.
Dada la complejidad de la cardiopatía congénita no pudo ser intervenida quirúrgicamente en nuestro hospital y, aunque se realizaron los trámites pertinentes para derivarle a otro centro cardiológico, no pudo ser transferida. La paciente falleció a los siete meses de edad. No se realizó autopsia por falta de consentimiento de los padres.
Discusión
La anoftalmía puede estar asociada con varios síndromes tales como el de Lenz, CHARGE (coloboma, cardiopatía –heart-, atresia de coanas, retraso psicomotor y del crecimiento, genitales anómalos, malformaciones auriculares –ear- y/o sordera), Fraser, Matthew-Wood o de Spear o anoftalmía/microftalmía-hipoplasia pulmonar-hernia diafragmática-cardiopatía, óculo-facio-cardio-dental y anoftalmía-esofágico-genital e isomerismo. Comprende anomalías genéticas tanto dominantes (mutaciones SOX2) como recesivas (mutaciones CHX10) o asociadas a infecciones durante el embarazo, como la causada por el citomegalovirus [2],[3],[9],[10]. En el caso presentado, la asociación de anoftalmía, malformaciones cardíacas tanto derechas como izquierdas e hipertensión pulmonar es muy rara y su etiología no ha sido dilucidada hasta el momento.
Al igual que en otros países de la región, en Ecuador también existe limitación para realizar estudios genéticos complejos, lo que constituye un reto al momento de dar un consejo genético a la familia. Debido a los antecedentes y a la consanguinidad existente entre los progenitores, se alertó a los padres acerca de la alta probabilidad de que un nuevo descendiente padezca de patologías similares, debido a un probable patrón hereditario.
A pesar del diagnóstico confirmado de hipertensión pulmonar en nuestra paciente, no se indicó medicación vasodilatadora pulmonar. Esto debido a que el riesgo de propiciar robo de flujo coronario al disminuir las resistencias vasculares pulmonares era alto, en una paciente con arteria coronaria izquierda originada anómalamente desde la arteria pulmonar. En consecuencia, el riesgo de desencadenar falla cardíaca aguda era mayor que el beneficio de la medicación.
Conclusiones
El espectro de asociaciones de anoftalmía y cardiopatías congénitas es diverso y amplio, existiendo varios síndromes ya ilustrados en los que estos fenotipos se asocian. Sin embargo, las cardiopatías congénitas complejas que se presentaron en esta paciente han sido previamente descritas como entidades separadas y no como parte de un sólo síndrome como el que hemos comunicado. El presente reporte adhiere a las directrices CARE para el reporte de casos clínicos [11].
Notas
Aspectos éticos
El consentimiento informado solicitado por Medwave, ha sido firmado por el padre de la paciente. Una copia de este fue remitida a la dirección editorial de la Revista.
Declaración de conflictos de intereses
Los autores han completado el formulario de declaración de conflictos intereses del ICMJE traducido al castellano por Medwave, y declaran no haber recibido financiamiento para la realización del reporte; no tener relaciones financieras con organizaciones que podrían tener intereses en el artículo publicado, en los últimos tres años; y no tener otras relaciones o actividades que podrían influir sobre el artículo publicado. Los formularios pueden ser solicitados contactando al autor responsable o a la dirección editorial de la Revista.
Financiamiento
Los autores declaran que no hubo fuentes de financiación externas.

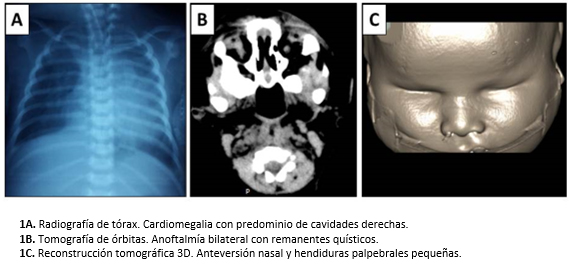 Figura 1. Exámenes de imagenología.
Figura 1. Exámenes de imagenología.
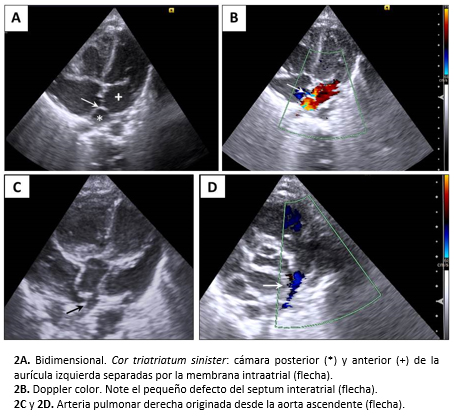 Figura 2. Ecocardiografía.
Figura 2. Ecocardiografía.
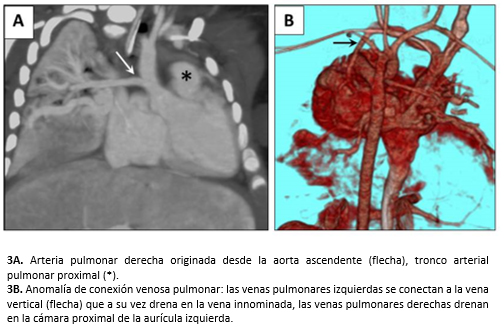 Figura 3. Angiotomografía cardíaca.
Figura 3. Angiotomografía cardíaca.
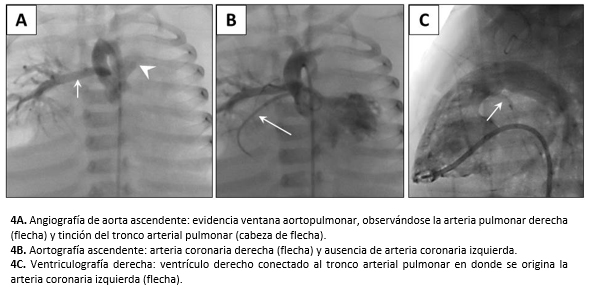 Figura 4. Cateterismo cardíaco.
Figura 4. Cateterismo cardíaco.
 Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave está bajo una licencia Creative Commons Atribución-NoComercial 3.0 Unported. Esta licencia permite el uso, distribución y reproducción del artículo en cualquier medio, siempre y cuando se otorgue el crédito correspondiente al autor del artículo y al medio en que se publica, en este caso, Medwave.
La anoftalmía congénita clínica es la ausencia uni o bilateral del globo ocular, que se presenta de forma aislada o como parte de un síndrome. Tiene muy baja prevalencia y su etiología es heterogénea. La presencia conjunta de malformaciones cardíacas congénitas complejas también es poco frecuente. La asociación de anoftalmía congénita con cardiopatías congénitas es más rara aún, la etiología de tales asociaciones no es bien comprendida todavía. Se reporta el caso de una paciente que presentó la muy infrecuente asociación de anoftalmía bilateral, múltiples malformaciones cardíacas e hipertensión arterial pulmonar grave.
Authors:
Raúl Enrique Ríos-Méndez [1 ], Michell Marola Lozano Chinga [1 ]
Affiliation:
[1] Cardiología/Neonatología, Hospital Pediátrico Baca Ortiz, Quito, Ecuador
E-mail: riosmendez@intramed.net.ar
Author address:
[1] Avenida Colón y 6 de Diciembre
Sin número
Quito
Ecuador
Citation: Ríos-Méndez RE , Lozano Chinga MM . Rare association of anophthalmia, complex congenital heart disease and pulmonary hypertension: case report. Medwave 2016 Oct;16(9):e6568 doi: 10.5867/medwave.2016.09.6568
Submission date: 18/6/2016
Acceptance date: 7/9/2016
Publication date: 7/10/2016
Origin: not requested
Type of review: reviewed by two external peer reviewers, double-blind
Comments (0)
We are pleased to have your comment on one of our articles. Your comment will be published as soon as it is posted. However, Medwave reserves the right to remove it later if the editors consider your comment to be: offensive in some sense, irrelevant, trivial, contains grammatical mistakes, contains political harangues, appears to be advertising, contains data from a particular person or suggests the need for changes in practice in terms of diagnostic, preventive or therapeutic interventions, if that evidence has not previously been published in a peer-reviewed journal.
No comments on this article.
To comment please log in
Medwave provides HTML and PDF download counts as well as other harvested interaction metrics. There may be a 48-hour delay for most recent metrics to be posted.
- Morrison D, FitzPatrick D, Hanson I, Williamson K, van Heyningen V, Fleck B, et al. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet. 2002 Jan;39(1):16-22. | PubMed |
- Verma AS, Fitzpatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007 Nov 26;2:47. | PubMed |
- Bermejo Sánchez E, Ayala Garcés A, Félix Rodríguez V, Martín Bermejo M, Blanco García M, Egüés Jimeno J, et al. [Anophthalmia/micro-ophthalmia in syndromes: epidemiology study of newborns in Spain]. An Esp Pediatr. 1996 Sep;45(3):269-75. | PubMed |
- Wang J, Steelman CK, Vincent R, Richburg D, Chang TS, Shehata BM. Two case reports of anophthalmia and congenital heart disease: Adding a new dimension to this association. Fetal Pediatr Pathol. 2010;29(5):291-8. | CrossRef | PubMed |
- Pasutto F, Sticht H, Hammersen G, Gillessen-Kaesbach G, Fitzpatrick DR, Nürnberg G, et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet. 2007 Mar;80(3):550-60. | PubMed |
- Segel R, Levy-Lahad E, Pasutto F, Picard E, Rauch A, Alterescu G, et al. Pulmonary hypoplasia-diaphragmatic hernia-nophthalmia-cardiac defect (PDAC) syndrome due to STRA6 mutations--what are the minimal criteria? Am J Med Genet A. 2009 Nov;149A(11):2457-63. | CrossRef | PubMed |
- Herman TE, Siegel MJ. Anopthalmia/microphthlamia-esophageal atresia association with additional features imperforate anus, choanal atresia, cyanotic heart disease. J Perinatol. 2012 Oct;32(10):814-6. | CrossRef | PubMed |
- Nakata S, Imai Y, Takanashi Y, Kurosawa H, Tezuka K, Nakazawa M, et al. A new method for the quantitative standardization of cross-sectional areas of the pulmonary arteries in congenital heart diseases with decreased pulmonary blood flow. J Thorac Cardiovasc Surg. 1984 Oct;88(4):610-9. | PubMed |
- McCarthy RW, Frenkel LD, Kollarits CR, Keys MP. Clinical anophthalmia associated with congenital cytomegalovirus infection. Am J Ophthalmol. 1980 Oct;90(4):558-61. | PubMed |
- Ragge NK, Subak-Sharpe ID, Collin JR. A practical guide to the management of anophthalmia and microphthalmia. Eye (Lond). 2007 Oct;21(10):1290-300. | PubMed |
- Case reports. A little structure goes a long way. care-statement.org [on line]. | Link |
Morrison D, FitzPatrick D, Hanson I, Williamson K, van Heyningen V, Fleck B, et al. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet. 2002 Jan;39(1):16-22. | PubMed |Verma AS, Fitzpatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007 Nov 26;2:47. | PubMed |Bermejo Sánchez E, Ayala Garcés A, Félix Rodríguez V, Martín Bermejo M, Blanco García M, Egüés Jimeno J, et al. [Anophthalmia/micro-ophthalmia in syndromes: epidemiology study of newborns in Spain]. An Esp Pediatr. 1996 Sep;45(3):269-75. | PubMed |Wang J, Steelman CK, Vincent R, Richburg D, Chang TS, Shehata BM. Two case reports of anophthalmia and congenital heart disease: Adding a new dimension to this association. Fetal Pediatr Pathol. 2010;29(5):291-8. | CrossRef | PubMed |Pasutto F, Sticht H, Hammersen G, Gillessen-Kaesbach G, Fitzpatrick DR, Nürnberg G, et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet. 2007 Mar;80(3):550-60. | PubMed |Segel R, Levy-Lahad E, Pasutto F, Picard E, Rauch A, Alterescu G, et al. Pulmonary hypoplasia-diaphragmatic hernia-nophthalmia-cardiac defect (PDAC) syndrome due to STRA6 mutations--what are the minimal criteria? Am J Med Genet A. 2009 Nov;149A(11):2457-63. | CrossRef | PubMed |Herman TE, Siegel MJ. Anopthalmia/microphthlamia-esophageal atresia association with additional features imperforate anus, choanal atresia, cyanotic heart disease. J Perinatol. 2012 Oct;32(10):814-6. | CrossRef | PubMed |Nakata S, Imai Y, Takanashi Y, Kurosawa H, Tezuka K, Nakazawa M, et al. A new method for the quantitative standardization of cross-sectional areas of the pulmonary arteries in congenital heart diseases with decreased pulmonary blood flow. J Thorac Cardiovasc Surg. 1984 Oct;88(4):610-9. | PubMed |McCarthy RW, Frenkel LD, Kollarits CR, Keys MP. Clinical anophthalmia associated with congenital cytomegalovirus infection. Am J Ophthalmol. 1980 Oct;90(4):558-61. | PubMed |Ragge NK, Subak-Sharpe ID, Collin JR. A practical guide to the management of anophthalmia and microphthalmia. Eye (Lond). 2007 Oct;21(10):1290-300. | PubMed |





 Research papers
Research papersSystematization of initiatives in sexual and reproductive health about good practices criteria in response to the COVID-19 pandemic in primary health care in Chile
Clinical, psychological, social, and family characterization of suicidal behavior in Chilean adolescents: a multiple correspondence analysis
