
Key Words: hereditary angioedema type I, complement C1 inhibitor protein, diagnoses
Abstract
Hereditary angioedema is a rare disease with great heterogeneity of symptoms such as edema of the skin, gastro-intestinal mucosa and larynx or pharynx. Even though there are three types, the most frequent is type I, which is a result from a deficiency of the complement C1 inhibitor. The severity of its symptoms along with the low prevalence of the disease and the need for appropriate specific treatment make the diagnosis and treatment of the pathology an outstanding subject for the family physician. The present is the case of a male teenager with alpha-1 antitrypsin deficiency since he was six months old, angioedema on arms and legs since 11 years old and diagnosed with hereditary angioedema type I one year after. The definitive diagnosis of the disease enabled an appropriate treatment which consists in preventing outbreaks that may compromise the patient's life and, if they occur, administration of complement C1 inhibitor.
Introduction
Hereditary angioedema (HAE) or Quincke´s edema (name given by its discoverer in 1882), is a genetic disease characterized by defined and deformational swelling involving skin, subcutaneous tissue, mucous membranes and sometimes viscera.
It is a rare disease, with low prevalence (between one and nine cases per 100,000 people) [1]. It presents with a wide range of clinical manifestations and is often erroneously diagnosed as an autoimmune disease or anaphylaxis. The development of glottis edema in 25 to 30% of cases [2] and 13% of deaths from asphyxia [3] justify the need of an early diagnosis and the implementation of a prophylactic treatment to reduce severe symptoms that can compromise patients´ life. In this case, the administration of the only valid treatment to prevent death by this cause: C1 esterase inhibitor (C1-inh). We find ourselves before an illness with a great impact in the quality of life of those who suffer it and their families because of both recurrence and the severity of the symptoms.
After reviewing the existing literature, we have found associations between hereditary angioedema and autoimmune diseases such as thyroiditis, lupus erythematosus, Sjögren´s syndrome or inflammatory bowel disease [4] but no other case associating alpha 1-antitrypsin deficiency with C1-inh deficiency.
Case report
The present is the case of a teenager, native of Cádiz (Spain) whose parents gave informed consent for this presentation.
Personal history of interest includes an alpha 1-antitrypsin deficiency made manifest when he was 9 months old after several episodes of bronchiolitis and follow-ups by Puerta del Mar university hospital (Cádiz). Alpha 1-antitrypsin values have been rising gradually with no need of specific treatment, ranging from 76.9mg/dl when the patient was one year old, to nearly normalizing at 14 years of age with 88.6mg/dl and no hepatic or gastrointestinal impact so far. Currently the young man suffers from non-allergic bronchial asthma with ongoing exacerbations that cause moderate effort dyspnea even following treatment with plusvent ®25/200mcg aerosol inhaler and montelukast 5mg. Current weight is 52kg and height is 156cm, his body mass index is 21.37 being at the 75th percentile.
Among his family history, his father also suffers alpha-1 antitrypsin deficiency following no treatment and his maternal grandmother´s cousin died of systemic lupus erythematosus. There are no evident backgrounds of recurrent angioedema on the maternal or paternal linage. Patient and parents are pending genetic study.
At 11 years of age he starts suffering recurrent angioedema of lower limbs, of pale reddish or whitish color, not associated to urticaria, painful to the touch and causing discomfort when appearing in joints. In several occasions he has experienced palpebral and facial edema, the first times repeating every 30 or 45 days, with no apparent cause. Nearly all cases were treated with steroids and antihistamines in both primary health care and hospital improving in 36 hours with or without treatment.
After a 10 month-course of evolution he suffers an episode of edema of tongue, uvula and pharynx and was treated as in other occasions. The outbreaks have increased to 2-3 a month.
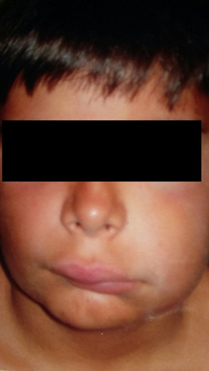
Figure 1. Facial angioedema
It was decided to make a dermatological and allergological study after persistence, increased frequency (up to 10 outbreaks in 6 months) and severity of the episodes, allergic diseases and anaphylactic reaction were discarded. This was demonstrated analytically determining immunoglobulin E (IgE) serum within normal levels.
Among the findings are:
- Serum total Ig E: 19.80 UI/ml
- Specific allergen Ig E: negative
- Prick-test with regular neumollergy provocation: negative
- Food prick-test: negative
- Latex prick-test: negative
- Simplex anisakis prick-test: negative
- ASLO: 184UI/ml
- TSH: 4.6mcU/ml
- FT4: 1.2mcg/ml
- ANA: negative
- Proteinogram: Albumin: 4.3g/dl; reference value 3.5-5.3
Alpha-1 globulins: 0.3g/dl; reference value 0.1-0.3
Alpha-2 globulins: 0.6g/dl; reference value 0.4-0.8
Beta-globulins: 0.7g/dl; reference value 0.5-1
Gamma-globulins 1.1g/dl; reference value 0.8-1.6 - Albumin/globulin ratio: 1.6 reference value 1.3-2.2
As consequence of the findings it was decided to expand the research with the following:
|
Values found |
Reference values |
|
|
Complement C3 |
148.20 mg/dl |
90-180 mg/dl |
|
Complement C4 |
0.90 mg/dl |
10-40 mg/dl |
|
Complement C1q |
22.30 mg/dl |
10-25 mg/dl |
|
C1 Esterase Inhibitor |
6.14 mg/dl |
22-34 mg/dl |
|
Complement C5 |
19.90 mg/dl |
4-15 mg/dl |
The clinical and laboratory data (low levels of C4, normal levels of C1q and C1 inh at 24% of its normal value) confirmed a diagnosis of hereditary angioedema type I, thus starting an individualized treatment appropriate for his pathology. From this moment on, the use of steroids, antihistamines and epinephrine for outbreaks was discarded. Currently, patient follow-up is carried out by the allergy service at Virgen del Rociío university hopital (Sevilla).
Fourteen months passed since the appearance of first symptoms to the diagnosis. Parents quantified approximately 15 outbreaks of angioedema over the period. The age of the patient (12 years old) at the time of diagnosis of hereditary angioedema, allowed to discard attenuated androgens as preventive treatment during outbreaks and antifibrinolytics were elected as long-term prophylaxis. The treatment consists of tranexamic acid 500 mg every 8 hours and has managed to decrease the frequency of outbreaks and intensity.
Since the onset of treatment 20 months ago, the frequency of outbreaks has decreased considerably, having had only three outbreaks consisting of edemas of smaller size on upper limbs and one episode of palpebral edema with no need for medical treatment and with no edemas in respiratory tract or digestive symptoms. One of the outbreaks was associated with a 48 hour fever whilst the other two had no apparent cause. None were over 24 hours. The patient was given Cinryze® (human C1 inh) for intravenous use in case of important acute outbreak or as short-term prophylaxis in case of surgical procedure, without need of use so far.
The preventive treatment with antifibrinolytics is well tolerated by patient but forces to kidney, hepatic and ophthalmological follow-ups to control retinopathy and glaucoma with no complications so far. Currently he maintains the same dose of tranexamic acid since the beginning of treatment.
The control of acute outbreaks since he started treatment made parents and patient decrease the negative perception of the disease and have a greater expectation of health.
Discussion
Angioedema received the name of angioneurotic edema for the first time in 1882, thanks to Heinrich Quincke. In 1888, William Osler spoke about a hereditary basis and in 1917 its autosomal dominant inheritance was identified. It was in 1963 when Donaldson and Evans described the genetic cause of the disease as a deficiency of the serum C1 complement (C1 inh) [5].
The genetic deficiency of angioedema can have a genetic background or can be acquired and is evidenced by the absence or insufficient synthesis of C1 inh [6], needed to regulate the activation of the complement system classical pathway. C1 inh belongs to the serine-protease inhibitor system (serpin) of human plasma and other proteins as ant thrombin III, alpha-2 antiplasmin, alpha-1 antitrypsin (whose deficiency is also found in our patient).
Classically, two types of hereditary angioedema have been described. Hereditary angioedema type I is the most common, representing about 85% of the cases. It is caused by a deficiency of synthesis of C1 inhibitor complement, as in the case of our teenager. Type II represents the remaining 15% and, even though the patient can present normal or even high values of C1 inh they are functionally deficient. Despite this classification a third variant has been described, hereditary angioedema type III [7],[8], with the same clinical manifestations as the two previous ones, with normal values and functionality of C1 inh. It only affects woman, estrogen dependent, related to pregnancy and use of contraceptives. There are only a few cases of this last type [9].
Hereditary angioedema is a rare disease, with low prevalence, between one and nine per 100,000 individuals, with no gender differences or predominance of race [1], except hereditary angioedema type III that appears among woman and is X-linked [5].
Despite typology of the disease, clinical manifestations are virtually identical in any of its variants. Such conditions include recurrent episodes self-limited in time of facial edema that affects subcutaneous tissue, viscera, mucous of gastrointestinal and upper airways [10] with high variability of symptoms. The severity depends on the degree of affectation and location of the edema. Usually cutaneous angioedema is not painful, not associated to urticaria, whitish and not red as described in this teenager. Other symptoms that can appear are edema of glottis and/or pharynx, dyspnea, dysphagia, abdominal distention, vomiting, diarrhea, and constipation, so far our patient has not had digestive symptoms. Certain circumstances such as stress, infectious procedures, surgery, intubation, invasive dental procedures [11], trigger acute outbreaks, so we can consider that the fever conditioned the exacerbation of the outbreak of the young man.
Hereditary angioedema should be suspected in recurring events of subcutaneous angioedema without urticaria, repeated episodes of self-limited abdominal pain or non-anaphylactic laryngeal edema. The diagnosis of the disease is confirmed by the serum complement levels [8],[12] like in our case, screening is easy:
- Low levels of C4 (<30%) makes us suspect angioedema.
- Normal levels of C1q suggest a hereditary angioedema, but if levels are low, it suggests an acquired angioedema.
- Low levels of C1 inh (<30% in two determinations spaced at least 1 month) in a hereditary angioedema suggest we have a hereditary angioedema type I.
- Normal or high levels of C1 inh talks about a hereditary angioedema type III.
The onset of the clinical manifestations can be at any age although it is mostly found in childhood and adolescence [4], as the case of our patient starting symptoms at 11 years of age. He had C1 inh 3.6 times under reference values and C4 four times below. These data confirm the diagnosis of hereditary angioedema type I.
To prevent complications it is important to start early treatment. The purposes of prophylactic and acute treatment are to reduce intensity of edemas, avoid mortality and avoid negative emotional impact [13], objectives achieved so far in this case passing from 15 episodes in a year to three episodes after stating treatment. This treatment includes:
- For long-term prevention there are two different treatments. Antifibrinolytics like tranexamic acid 20-50 mg/kg/day are used in children (as used in this case) or aminocaproic acid up to 16mg/day [11]. In adults attenuated androgens like danazol 600 mg per day are used. The possible side-effects and adverse effects can make treatments change regardless of age [5], in our case initial treatment is well tolerated with no complications.
- If there is an acute outbreak with risk to the patient (airway compromise or severe abdominal condition) an intravenous concentrate of C1 inh derived from human plasma, 500–1000 UI (Berinert® and Cinryze®) should be administrated. The patient was given Cinryze® 1000 UI to administrate half of a vial if necessary (considering that his weight id 52kg), it can be repeated 30 minutes after if necessary.
- In case of surgery, intubation, tooth removal or any other procedures that involves trauma in the cervicofacial area, a possible acute outbreak should be prevented using C1 inh concentrate thirty minutes before [12] invasive procedure or if not, attenuated androgens several days before.
We are facing a disease which, even though 75% of the patients have their first outbreak before they are 15 years old, it takes up to 8 [14] and 15 [3] years to diagnose. This has a particular importance because at the present mortality from hereditary angioedema drastically reduces once diagnosed and correct treatment is started.
A well led differential diagnosis is important because digestive symptoms can confuse medical staff with an acute abdomen. Pharyngeal and laryngeal edema can be treated like an anaphylactic reaction. This situation is quite serious because hereditary angioedema does not respond to steroids, antihistamines or adrenalin, and can cause death from asphyxia with a mortality between 13 and 30% [3],[4]. Besides, the patient has an alpha-1 antitrypsin deficiency as an underlying pathology and in literature reviewed; we did not find any description of comorbidity associated to hereditary angioedema. Apparently, this seems to be the first case report; however, there are reports in the last years of associations between hereditary angioedema and autoimmune diseases like systemic lupus erythematosus, a disease found in a family member.
Conclusion
Even though hereditary angioedema is a disease with low prevalence, it can be mortal if not diagnosed on time because the clinical picture can make us think about other pathologies thus treating patients erroneously. Our patient suffered at least 15 outbreaks diagnosed as urticaria or anaphylactic reaction at the emergency rooms of both primary health care and hospital until correct diagnosis. This information leads us to the importance of reviewing this disease not well known among medical staff that can be seen in primary health care, entry point of all diseases, and easy to diagnose if when we suspect angioedema we decide to determinate serum complement values.
Notes
From the editor
The authors originally submitted this article in Spanish and subsequently translated it into English. The Journal has not copyedited this version.
Ethical aspects
The patient's mother signed the informed consent form requested by Medwave. A copy of this document was sent to the editorial direction of the Journal.
Conflicts of interest
The authors completed the ICMJE declaration of conflicts of interest, translated into Spanish by Medwave, and declare they have not received funding for the completion of the report; have no financial relationships with organizations that may have interests in the article published in the last three years; and have no other relationships or activities that could influence the published article. Forms can be requested by contacting the responsible author or editorial direction of the Journal.
Funding
The authors declare there are no external funding sources for this article.
 Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.
Esta obra de Medwave estß bajo una licencia Creative Commons Atribuci¾n-NoComercial 3.0 Unported. Esta licencia permite el uso, distribuci¾n y reproducci¾n del artĒculo en cualquier medio, siempre y cuando se otorgue el crķdito correspondiente al autor del artĒculo y al medio en que se publica, en este caso, Medwave.

El angioedema hereditario es una enfermedad rara, de gran heterogeneidad en los sĒntomas, manifestßndose con edema a nivel cutßneo, mucosa gastrointestinal y de laringe/faringe. Aunque existen tres variedades, el tipo I es el mßs frecuente y es provocado por una deficiencia en la sĒntesis del complemento C1 inhibidor. La gravedad de la clĒnica, junto a la baja prevalencia de la enfermedad y la necesidad de un tratamiento especĒfico, hacen que el diagn¾stico y tratamiento de dicha patologĒa sea a·n una asignatura pendiente para el mķdico de familia en atenci¾n primaria. Presentamos el caso de un adolescente var¾n con dķficit de α-1 antitripsina desde los seis meses de edad, con aparici¾n de angioedemas en piernas y brazos a los 11 a±os, diagnosticado de angioedema hereditario tipo I un a±o despuķs. El diagn¾stico definitivo de la enfermedad permiti¾ instaurar un tratamiento adecuado a su patologĒa, que consiste en la prevenci¾n de brotes que puedan comprometer la vida del paciente y, en el caso de que aparezcan, en la administraci¾n del complemento C1 inhibidor.
Authors:
Francisca Mu±oz Peralta [1], Eva Buller Vigueira [2 ], Juana Cabello Pulido [3]
Affiliation:
[1] Servicio Andaluz de Salud, Centro de Salud Gonzalo Pķrez Fabra, Paterna de Rivera, Cßdiz, Espa±a
[2] Servicio Andaluz de Salud, Centro de Salud Virgen de la Frontera, Vejer de la Frontera, Cßdiz, Espa±a
[3] Servicio Andaluz de Salud, Centro de Salud RodrĒguez Arias, San Fernando, Cßdiz, Espa±a
E-mail: pakimupe@outlook.com
Author address:
[1] Centro de Salud Gonzalo Pķrez Fabra
Calle Molinos 14
Paterna de Rivera
Cßdiz
Espa±a
Citation: Mu±oz Peralta F , Buller Vigueira E, Cabello Pulido J . Hereditary angioedema type I: a case report. Medwave 2016 Ene;16(1):e6378 doi: 10.5867/medwave.2016.01.6378
Submission date: 4/11/2015
Acceptance date: 18/12/2016
Publication date: 28/1/2016
Origin: not requested
Type of review: reviewed by one external peer reviewer, double-blind
Comments (0)
We are pleased to have your comment on one of our articles. Your comment will be published as soon as it is posted. However, Medwave reserves the right to remove it later if the editors consider your comment to be: offensive in some sense, irrelevant, trivial, contains grammatical mistakes, contains political harangues, appears to be advertising, contains data from a particular person or suggests the need for changes in practice in terms of diagnostic, preventive or therapeutic interventions, if that evidence has not previously been published in a peer-reviewed journal.
No comments on this article.
To comment please log in
Medwave provides HTML and PDF download counts as well as other harvested interaction metrics. There may be a 48-hour delay for most recent metrics to be posted.
- August. Hereditary angioedema, 2011. Orpha.net [on line]. | Link |
- Jimenez Saab N, G¾mez Vera J, G¾mez Tiro J, Nieto MartĒnez S, Pliego Reyes C. Angioedema hereditario. Comunicaci¾n de un caso y revisi¾n de la bibliografĒa. Rev Aler Mķx. 2006; 53(1):34-41. | Link |
- Fernßndez Romero DS, Dimarco P, Malbran A. Angioedema hereditario. Historia familiar y manifestaciones clĒnicas en 58 pacientes. Medicina (Buenos Aires) 2009;69:601-606. | Link |
- Vives Toledo R, SorlĒ Guerola JV, Sierra Santos L, GarcĒa Ribes M. Angioedema hereditario. Rev ClĒn Med Fam. 2015; Vol 8(1):62-65. | Link |
- Navarro Ruiz A, Crespo C, Poveda Andrķs JL, Cebollero de Torre A. Algoritmo de diagn¾stico y tratamiento del angioedema hereditario como herramienta para su manejo. Farm Hosp. 2013;37(6):521-529. | CrossRef |
- Donalson VH, Evans RR. A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C1-esterase. Am J Med. 1963 Jul;35:37-44. | PubMed |
- Bork K, Barnstedt SE, Kock P, Traupe H. Hereditary Angioedema with normal inhibidor C1 esterasaibitor activity in women. Lancet. 2000 Jul 15;356(9225):213-7. | PubMed |
- ┴lvarez Caro F, DĒaz MartĒn JJ, ┴lvarez Berciano F. Edema angioneur¾tico hereditario en pediatrĒa: consideraciones a prop¾sito de tres casos. Act Ped Esp. 2007;65(6):300-303. | Link |
- Calvo G¾mez-Rodulfo A, GarcĒa L¾pez JE, Herrero-MorĒn JD, RodrĒguez GarcĒa G, Gonzßlez Guerra F. Angioedema hereditario en pediatrĒa. Bol Pediatr. 2009; 49:16-23. | Link |
- Past¾ Cardona L, Bordas Orpinell J, Mercadal Orfila G, Pķrez de la Vera A, J¾dar Massanķs R. Profilaxis y tratamiento del angioedema hereditario y adquirido en el HUB; utilizaci¾n del inhibidor de la C1-esterasa. Farm Hosp. 2003;27(6):346-352. | Link |
- Malbran A, Fernßndez Romero DS, Menķndez A. Angioedema hereditario. GuĒa de tratamiento. Medicina (Buenos Aires) 2012;72:119-123. | Link |
- Caballero T1, Baeza ML, Caba±as R, Campos A, Cimbollek S, G¾mez-Traseira C, et al. Consensus Statement on the Diagnosis, Management, and Treatment of Angioedema Mediated by Bradykinin. Part II. Treatment, Follow-up, and Special Situations. J Investig Allergol Clin Immunol. 2011;21(6):422-41; quiz 442-3. | PubMed |
- Rodriguez Labrada M, Surday Berm·dez L, Corrales ┴lvarez M. Enfermedad de Quincke, desafĒo del especialista en otorrinolaringologĒa. Acta Mķdica del Centro. 2014; Vol 8(3). | Link |
- Huang SW. Result of an online survey of patients with hereditary Angioedema. Allergy Asthma Proc. Allergy Asthma Proc. 2004 Mar-Apr;25(2):127-31. | PubMed |
Jimenez Saab N, G¾mez Vera J, G¾mez Tiro J, Nieto MartĒnez S, Pliego Reyes C. Angioedema hereditario. Comunicaci¾n de un caso y revisi¾n de la bibliografĒa. Rev Aler Mķx. 2006; 53(1):34-41. | Link |Fernßndez Romero DS, Dimarco P, Malbran A. Angioedema hereditario. Historia familiar y manifestaciones clĒnicas en 58 pacientes. Medicina (Buenos Aires) 2009;69:601-606. | Link |Vives Toledo R, SorlĒ Guerola JV, Sierra Santos L, GarcĒa Ribes M. Angioedema hereditario. Rev ClĒn Med Fam. 2015; Vol 8(1):62-65. | Link |Navarro Ruiz A, Crespo C, Poveda Andrķs JL, Cebollero de Torre A. Algoritmo de diagn¾stico y tratamiento del angioedema hereditario como herramienta para su manejo. Farm Hosp. 2013;37(6):521-529. | CrossRef |Donalson VH, Evans RR. A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C1-esterase. Am J Med. 1963 Jul;35:37-44. | PubMed |Bork K, Barnstedt SE, Kock P, Traupe H. Hereditary Angioedema with normal inhibidor C1 esterasaibitor activity in women. Lancet. 2000 Jul 15;356(9225):213-7. | PubMed |┴lvarez Caro F, DĒaz MartĒn JJ, ┴lvarez Berciano F. Edema angioneur¾tico hereditario en pediatrĒa: consideraciones a prop¾sito de tres casos. Act Ped Esp. 2007;65(6):300-303. | Link |Calvo G¾mez-Rodulfo A, GarcĒa L¾pez JE, Herrero-MorĒn JD, RodrĒguez GarcĒa G, Gonzßlez Guerra F. Angioedema hereditario en pediatrĒa. Bol Pediatr. 2009; 49:16-23. | Link |Past¾ Cardona L, Bordas Orpinell J, Mercadal Orfila G, Pķrez de la Vera A, J¾dar Massanķs R. Profilaxis y tratamiento del angioedema hereditario y adquirido en el HUB; utilizaci¾n del inhibidor de la C1-esterasa. Farm Hosp. 2003;27(6):346-352. | Link |Malbran A, Fernßndez Romero DS, Menķndez A. Angioedema hereditario. GuĒa de tratamiento. Medicina (Buenos Aires) 2012;72:119-123. | Link |Caballero T1, Baeza ML, Caba±as R, Campos A, Cimbollek S, G¾mez-Traseira C, et al. Consensus Statement on the Diagnosis, Management, and Treatment of Angioedema Mediated by Bradykinin. Part II. Treatment, Follow-up, and Special Situations. J Investig Allergol Clin Immunol. 2011;21(6):422-41; quiz 442-3. | PubMed |Rodriguez Labrada M, Surday Berm·dez L, Corrales ┴lvarez M. Enfermedad de Quincke, desafĒo del especialista en otorrinolaringologĒa. Acta Mķdica del Centro. 2014; Vol 8(3). | Link |Huang SW. Result of an online survey of patients with hereditary Angioedema. Allergy Asthma Proc. Allergy Asthma Proc. 2004 Mar-Apr;25(2):127-31. | PubMed |





 Research papers
Research papersSystematization of initiatives in sexual and reproductive health about good practices criteria in response to the COVID-19 pandemic in primary health care in Chile
Clinical, psychological, social, and family characterization of suicidal behavior in Chilean adolescents: a multiple correspondence analysis
